Chapter: Biotechnology Applying the Genetic Revolution: Environmental Biotechnology
Sequence Dependent Techniques for Metagenomics
SEQUENCE-DEPENDENT
TECHNIQUES FOR METAGENOMICS
Historically, sequence-based
metagenomics techniques were performed directly on samples from the environment
after culturing. In 1985, Pace and colleagues directly sequenced the 5S and 16S
rRNA gene sequences from the environment without culturing. This was
technically difficult at the time. After PCR techniques became more prevalent
in the early 1990s, the analyses of specific groups of organisms became
simpler. Researchers used PCR primers specific to 16S rRNA sequences to
identify the different organisms within the environmental sample. Yet these
techniques rely on some prior knowledge of the sequences. Moreover, this method
identifies a new organism only by its 16S rRNA sequence. It does not reveal the
physiology or genetics of the organism.
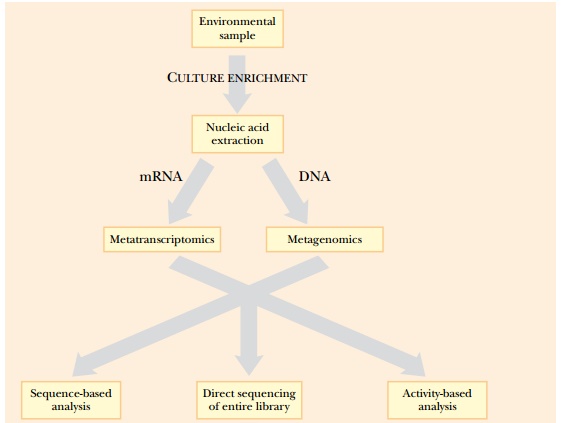
Making and using metagenomic libraries is a way to
identify the entire genetic complement of newly discovered life forms, without
culturing them. From the genome sequences the physiology and function of the
organism can be determined. DNA (or RNA) from the environment is isolated (and
in some cases may be enriched as described earlier). The DNA, which is a
mixture of fragments from multiple genomes, is then made into a library. Such
libraries are like traditional genomic libraries, except that the starting DNA
has sequences from many different organisms rather than one.
Basically, the total DNA
isolated from an environmental sample is cloned into a vector plasmid, and
constructs are maintained in Escherichia
coli. Of course, all the same library permutations apply as for a single
genome library. Large DNA fragments can be cloned into BACs (bacterial
artificial chromosomes), and small pieces can be cloned into plasmids. The
environmental DNA can be cloned into various expression vectors to determine if
any specific enzyme or protein is present in the library. Environmental mRNA
can also be isolated and converted to cDNA using reverse transcriptase before
cloning. Such a cDNA library will represent genes that are being actively
expressed in the environment. This approach circumvents the cloning of
noncoding DNA. Of course, the
same caveat exists, that is,
the library is only as good as its original DNA or mRNA. If these are
contaminated or sheared into small pieces, the library is worthless. In
addition, some DNA from the environment may not be represented in the library
because it cannot be isolated.
Metagenomic libraries can be sequenced to identify new genes or can be screened for new protein functions (Fig. 12.4). Many environmental biologists want to develop or find new enzymes that degrade pollutants or contaminants. Screening the library with a sequence containing conserved domains of known enzymes can find new enzymes. Along the same lines, PCR primers to conserved domains of known enzymes can amplify never-before-seen genes from the library. For example, aromatic oxygenases facilitate the degradation of aromatic hydrocarbons found in oil and coal. These contaminants can be degraded by a variety of different organisms. The genes and pathways involved are key targets for pathway engineering. Using PCR primers to conserved regions of known aromatic oxygenases amplifies the genes for novel (but related) oxygenases found in the environment.
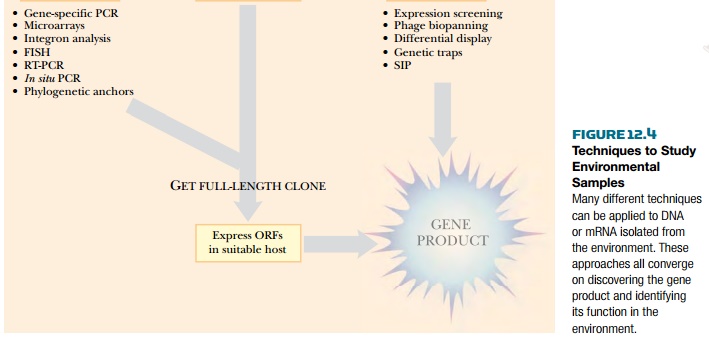
Some sequence-based analyses
for metagenomic libraries include microarrays, FISH, RT-PCR, sequencing using
phylogenetic anchors, and integron analysis (see later discussion). Sequencing
with phylogenetic anchors begins by identifying the sequence of a known gene. Often
a marker gene such as 16S rRNA gene is identified first, and then the regions
upstream or downstream of the marker are sequenced.
For example, a 16S rRNA
sequence from seawater classified a particular genomic fragment to the γ-Proteobacteria. Adjacent to
the 16S rRNA gene was a gene similar to bacteriorhodopsin, a transmembrane
proton pump that responds to light. The genes for bacteriorhodopsin
were originally thought to
exist only in the Archaea, but this analysis revealed that other inhabitants of
the ocean had similar genes.
Microarrays can also be used
to identify the types and numbers of different organisms in an environmental
sample. First, a microarray of unique sequences from known organisms is
created, and then fluorescently labeled environmental DNA is hybridized to the
array. The results can confirm whether or not a particular bacterium, virus, or
gene creature is present, and the relative abundance can be determined by the
intensity of fluorescence. FISH and RT-PCR can yield similar information, but
the environmental DNA is analyzed with a only few probes or primers,
respectively. The results are much more direct and focus on identifying known
organisms.
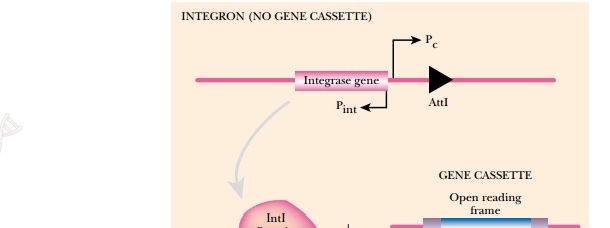
Integron analysis identifies open reading frames that are used by integrons and can identify more novel unknown genes than many previous techniques (Fig. 12.5). Integrons are related to transposons, but are particularly important for the spread of genes for antibiotic resistance and other properties that give the host a growth advantage in a particular environment. Integrons are genetic elements that contain a site (attI) to integrate a segment of DNA known as a gene cassette, a promoter to express the gene cassette (Pc ), and a gene for integrase (intI), the enzyme that recombines the gene cassette into the integron. Gene cassettes are segments of DNA with one or two open reading frames (ORFs) that lack promoters and are flanked by 59-base elements (59-be, also known as attC sites). When the integrase recognizes the 59-be sites, it excises the gene cassette and integrates it downstream of the Pc promoter. This allows the open reading frame to be expressed into protein. The 59-be sequence may vary in length, but must contain a conserved, seven-nucleotide sequence.
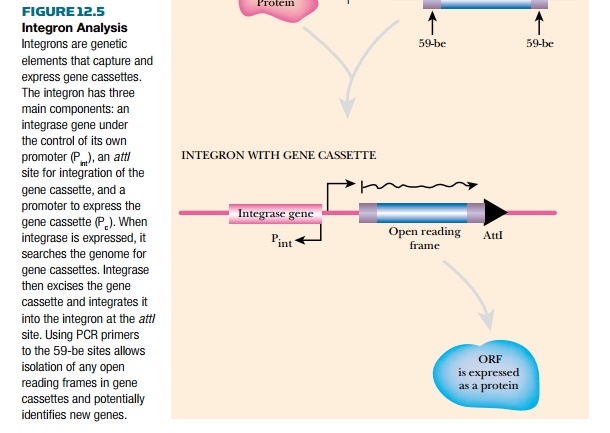
The gene cassettes are the
interesting part of the scenario and the key to integron analysis. Gene
cassettes were first identified because many encode antibiotic resistance
genes, but they may encode any type of gene. Screening metagenomic libraries
using PCR primers that recognize the 59-be elements amplifies these open
reading frames. This approach has identified novel genes related to DNA
glycosylases, phosphotransferases, and methyl transferases. Additionally, new
antibiotic resistance genes may be identified. Integron analysis is also useful
to study bacterial evolution and gene transfer because these elements can pass
from bacterium to bacterium during conjugation.
Related Topics