Chapter: Basic & Clinical Pharmacology : Antimycobacterial Drugs
Second-Line Drugs for Tuberculosis
SECOND-LINE DRUGS FOR
TUBERCULOSIS
The
alternative drugs listed below are usually considered onlyin case of resistance
to first-line agents; (2) in case of failure of clinical response to
conventional therapy; and (3) in case of serious treatment-limiting adverse
drug reactions. Expert guidance to deal with the toxic effects of these
second-line drugs is desirable. For many drugs listed in the following text,
the dosage, emergence of resistance, and long-term toxicity have not been fully
established.
Ethionamide
Ethionamide
is chemically related to isoniazid and similarly blocks the synthesis of
mycolic acids. It is poorly water soluble and avail-able only in oral form. It
is metabolized by the liver.

Most
tubercle bacilli are inhibited in vitro by ethionamide, 2.5 mcg/mL or less.
Some other species of mycobacteria also are inhibited by ethionamide, 10
mcg/mL. Serum concentrations in plasma and tissues of approximately 20 mcg/mL
are achieved by a dosage of 1 g/d. Cerebrospinal fluid concentrations are equal
to those in serum.Ethionamide is administered at an initial dose of 250 mg once
daily, which is increased in 250-mg increments to the recom-mended dosage of 1
g/d (or 15 mg/kg/d), if possible. The 1 g/d dosage, though theoretically
desirable, is poorly tolerated because of the intense gastric irritation and
neurologic symptoms that commonly occur, and one often must settle for a total
daily dose of 500–750 mg. Ethionamide is also hepatotoxic. Neurologic symptoms
may be alleviated by pyridoxine.Resistance to ethionamide as a single agent
develops rapidly in vitro and in vivo. There can be low-level cross-resistance
between isoniazid and ethionamide.
Capreomycin
Capreomycin is a
peptide protein synthesis inhibitor antibiotic obtained from Streptomyces capreolus. Daily injection
of 1 g intra-muscularly results in blood levels of 10 mcg/mL or more. Such
concentrations in vitro are inhibitory for many mycobacteria, including
multidrug-resistant strains of M
tuberculosis.
Capreomycin (15
mg/kg/d) is an important injectable agent for treatment of drug-resistant
tuberculosis. Strains of M tuberculosis
that are resistant to streptomycin or amikacin usually are suscep-tible to
capreomycin. Resistance to capreomycin, when it occurs, may be due to an rrs mutation.
Capreomycin
is nephrotoxic and ototoxic. Tinnitus, deafness, and vestibular disturbances
occur. The injection causes significant local pain, and sterile abscesses may
occur.
Dosing of capreomycin
is the same as that of streptomycin. Toxicity is reduced if 1 g is given two or
three times weekly after an initial response has been achieved with a daily
dosing schedule.
Cycloserine
Cycloserine
is an inhibitor of cell wall synthesis. Concentrations of 15–20 mcg/mL inhibit
many strains of M tuberculosis. The
dosage of cycloserine in tuberculosis is 0.5–1 g/d in two divided oral doses.
Cycloserine is cleared renally, and the dose should be reduced by half if
creatinine clear-ance is less than 50 mL/min.
The
most serious toxic effects are peripheral neuropathy and central nervous system
dysfunction, including depression and psy-chotic reactions. Pyridoxine, 150
mg/d, should be given with cycloserine because this ameliorates neurologic
toxicity. Adverse effects, which are most common during the first 2 weeks of
therapy, occur in 25% or more of patients, especially at higher doses. Adverse
effects can be minimized by monitoring peak serum concentrations. The peak
concentration is reached 2–4 hours after dosing. The recommended range of peak
concentrations is 20–40 mcg/mL.
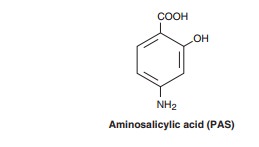
Aminosalicylic Acid (PAS)
Aminosalicylic
acid is a folate synthesis antagonist that is active almost exclusively against
M tuberculosis. It is structurally
similar to p-amino-benzoic acid
(PABA) and to the sulfonamides .
Tubercle
bacilli are usually inhibited in vitro by aminosalicylic acid, 1–5 mcg/mL.
Aminosalicylic acid is readily absorbed fromthe gastrointestinal tract. Serum
levels are 50 mcg/mL or more after a 4-g oral dose. The dosage is 8–12 g/d
orally for adults and 300 mg/kg/d for children. The drug is widely distributed
in tissues and body fluids except the cerebrospinal fluid. Aminosalicylic acid
is rapidly excreted in the urine, in part as active aminosalicylic acid and in
part as the acetylated compound and other metabolic prod-ucts. Very high
concentrations of aminosalicylic acid are reached in the urine, which can
result in crystalluria.
Aminosalicylic
acid is used infrequently because other oral drugs are better tolerated.
Gastrointestinal symptoms are common and may be diminished by giving the drug
with meals and with antacids. Peptic ulceration and hemorrhage may occur.
Hypersensitivity reactions manifested by fever, joint pains, skin rashes,
hepatosple-nomegaly, hepatitis, adenopathy, and granulocytopenia often occur
after 3–8 weeks of aminosalicylic acid therapy, making it necessary to stop
aminosalicylic acid administration temporarily or permanently.
Kanamycin & Amikacin
Kanamycin has been
used for treatment of tuberculosis caused by streptomycin-resistant strains,
but the availability of less toxic alter-natives (eg, capreomycin and amikacin)
has rendered it obsolete.
The role of amikacin
in treatment of tuberculosis has increased with the increasing incidence and
prevalence of multidrug-resistant tuberculosis. Prevalence of
amikacin-resis-tant strains is low (< 5%), and most multidrug-resistant strains
remain amikacin-susceptible. M
tuberculosis is inhibited at con-centrations of 1 mcg/mL or less. Amikacin
is also active against atypical mycobacteria. There is no cross-resistance
between strep-tomycin and amikacin, but kanamycin resistance often indicates
resistance to amikacin as well. Serum concentrations of 30–50 mcg/mL are
achieved 30–60 minutes after a 15 mg/kg intravenous infusion. Amikacin is
indicated for treatment of tuberculosis sus-pected or known to be caused by streptomycin-resistant
or multi-drug-resistant strains. Amikacin must be used in combination with at
least one and preferably two or three other drugs to which the isolate is
susceptible for treatment of drug-resistant cases. The recommended dosages are
the same as those for streptomycin.
Fluoroquinolones
In
addition to their activity against many gram-positive and gram-negative
bacteria, ciprofloxacin, levofloxa-cin, gatifloxacin, and moxifloxacin inhibit
strains of M tuberculosis at
concentrations less than 2 mcg/mL. They are also active against atypical
mycobacteria. Moxifloxacin is the most active against Mtuberculosis by weight in vitro. Levofloxacin tends to be slightly
moreactive than ciprofloxacin against M
tuberculosis, whereas ciprofloxa-cin is slightly more active against
atypical mycobacteria.
Fluoroquinolones
are an important addition to the drugs avail-able for tuberculosis, especially
for strains that are resistant to first-line agents. Resistance, which may
result from any one of several single point mutations in the gyrase A subunit,
develops rapidly if a fluoroquinolone is used as a single agent; thus, the drug
must be used in combination with two or more other active agents. Thestandard
dosage of ciprofloxacin is 750 mg orally twice a day. The dosage of
levofloxacin is 500–750 mg once a day. The dosage of moxifloxacin is 400 mg
once a day.
Linezolid
Linezolid inhibits
strains of M tuberculosis in vitro at
concentrations of 4–8 mcg/mL. It achieves good intra-cellular concentrations,
and it is active in murine models of tuberculosis. Linezolid has been used in
combination with other second- and third-line drugs to treat patients with
tuberculosis caused by multidrug-resistant strains. Conversion of sputum
cultures to negative was associated with linezolid use in these cases, and some
may have been cured. Significant and at times treatment-limiting adverse
effects, including bone marrow sup-pression and irreversible peripheral and
optic neuropathy, have been reported with the prolonged courses of therapy that
are necessary for treatment of tuberculosis. A 600-mg (adult) dose administered
once a day (half of that used for treatment of other bacterial infections)
seems to be sufficient and may limit the occurrence of these adverse effects.
Although linezolid may even-tually prove to be an important new agent for
treatment of tuber-culosis, at this point it should be considered a drug of
last resort for infection caused by multidrug-resistant strains that also are
resistant to several other first- and second-line agents.
Rifabutin
Rifabutin is derived
from rifamycin and is related to rifampin. It has significant activity against M tuberculosis, MAC, and Mycobacterium fortuitum . Its activity
is similar to thatof rifampin, and cross-resistance with rifampin is virtually
com-plete. Some rifampin-resistant strains may appear susceptible to rifabutin
in vitro, but a clinical response is unlikely because the molecular basis of
resistance, rpoB mutation, is the
same. Rifabutin is both substrate and inducer of cytochrome P450 enzymes.
Because it is a less potent inducer, rifabutin is indicated in place of
rifampin for treatment of tuberculosis in patients with HIV infec-tion who are
receiving antiretroviral therapy with a protease inhibitor or with a nonnucleoside
reverse transcriptase inhibitor (eg, efavirenz), drugs that also are cytochrome
P450 substrates.
The typical dosage of
rifabutin is 300 mg/d unless the patient is receiving a protease inhibitor, in
which case the dosage should be reduced to 150 mg/d. If efavirenz (also a
cytochrome P450 inducer) is used, the recommended dosage of rifabutin is 450
mg/d.
Rifabutin is effective
in prevention and treatment of dissemi-nated atypical mycobacterial infection
in AIDS patients with CD4 counts below 50/μL. It is also effective for
preventive therapy of tuberculosis, either alone in a 3–4 month regimen or with
pyrazi-namide in a 2-month regimen.
Rifapentine
Rifapentine is an
analog of rifampin. It is active against both M tuberculosis and MAC. As with all rifamycins, it is a
bacterialRNA polymerase inhibitor, and cross-resistance between rifampin and
rifapentine is complete. Like rifampin, rifapentine is a potent inducer
of cytochrome P450 enzymes, and it has the same drug interaction profile.
Toxicity is similar to that of rifampin. Rifapentine and its microbiologically
active metabolite, 25-desacetylrifapentine, have an elimination half-life of 13
hours. Rifapentine, 600 mg (10 mg/kg) once weekly, is indicated for treatment
of tuberculosis caused by rifampin-susceptible strains during the continuation
phase only (ie, after the first 2 months of therapy and ideally after
conversion of sputum cultures to negative). Rifapentine should not be used to
treat patients with HIV infec-tion because of an unacceptably high relapse rate
with rifampin-resistant organisms.
Related Topics