Chapter: Medical Microbiology: An Introduction to Infectious Diseases: Retroviruses, Human Immunodeficiency Virus, and Acquired Immunodeficiency Syndrome
Retroviruses - Virology
RETROVIRUSES
VIROLOGY
STRUCTURE
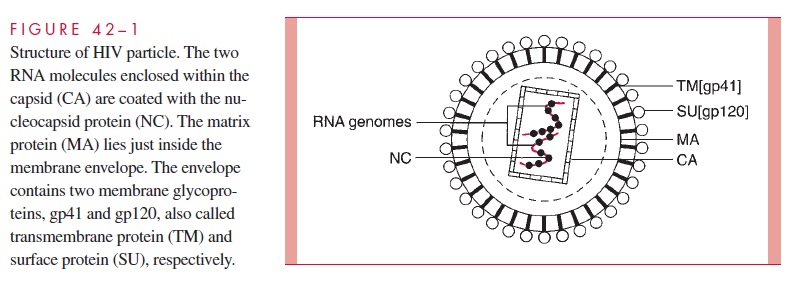
All retroviruses are remarkably similar in their basic composition. The structure of HIV-1 is depicted in Fig 42 – 1. The virion is about 100 nm in diameter, and because it contains two copies of the RNA genome, it is diploid. The RNA genome is coated with the nucleo-capsid protein (NC), and the RNA – protein complexes are enclosed in a capsid (CA) com-posed of multiple subunits. Like all enveloped viruses, the membrane is acquired during budding from the host cell, but the surface (SU, also called gp120) and transmembrane (TM, also called gp41) glycoproteins found in the envelope are virally encoded. Between the capsid and the envelope is the matrix (MA) protein. In addition to the structural pro-teins shown in Fig 42 – 1, the virion core contains three virus-specific proteins that are essential for viral replication: reverse transcriptase (RT), protease (PR), and integrase (IN). The relationships between the viral genes found in all retroviruses (gag, pol, and env) and the proteins they encode are presented in Table 42 – 1. Some retroviruses, including HIV-1, encode additional regulatory and accessory proteins as will be described below.
LIFE CYCLE
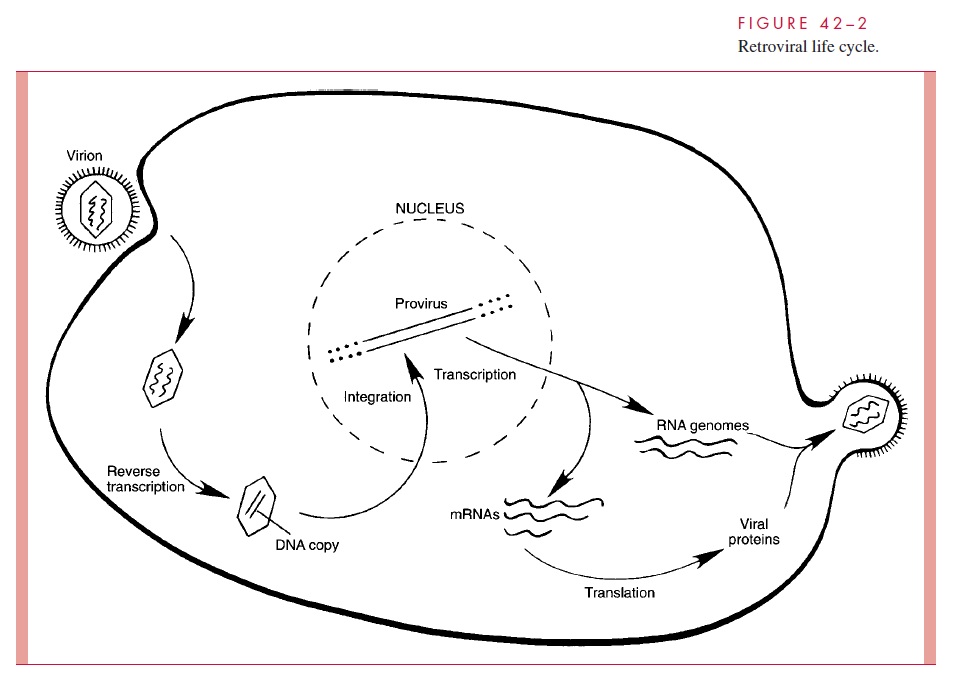
Figure 42 – 2 depicts the life cycle of a typical retrovirus and serves to illustrate the many unique aspects of retroviral replication that could be potential targets of therapeutic inter-vention.
Viral Entry
The virions adsorb to cellular membrane receptors and enter the cell by direct fusion with the plasma membrane. For HIV-1, the virion attachment protein is the SU glycoprotein gp120, and the cellular receptor is the CD4 molecule with one of the chemokine recep-tors, CXCR4 or CCR5 acting as coreceptors.
These receptors occur primarily in the plasma membrane of CD4 T lymphocytes, cells of the monocyte – macrophage series, and some other target cells. Early in the infection of an individual, the viruses are often macrophage-tropic because they preferentially use the CCR5 coreceptor. The emergence of syncytia-forming variants that use the CXCR4 coreceptor and are T-cell tropic appears to correlate with rapid advancement to AIDS. The HIV-1 transmembrane TM protein gp41 is responsible for fusion of the viral and cell membranes, leading to entry of the virion core complex into the cytoplasm of the cell.
HIV-1 can also infect cells such as fibroblasts and certain brain cells that lack the CD4 surface molecule, apparently because the chemokine receptors in combination with the fusion-inducing activity of the TM protein is sufficient in these cases to promote en-try. Fusion activity may also play an important role in amplification of the effects of the virus infection, particularly during the later stages of the infection, because infected cells expressing viral glycoproteins in their membranes readily fuse with uninfected CD4+ T lymphocytes to form large syncytia. This process appears to provide a means for cell-to-cell transmission of the virus that bypasses the usual extracellular phase and may con-tribute to the overall depletion of CD4+ lymphocytes in an infected individual.
Viral RNA Replication
Among the RNA viruses, retroviral replication is unique. Soon after entry of the viral core into the cytoplasm of the infected cell, the RNA is copied into double-stranded DNA by reverse transcriptase, the virion-associated DNA polymerase. The overall process is re-ferred to as reverse transcription and results in a linear DNA molecule that enters the nu-cleus and integrates more-or-less at random into a host cell chromosome. Once the viral genetic information has been converted to DNA and integrated, it essentially becomes part of the cellular genome. The viral genes, called the provirus, are therefore replicated and faithfully inherited as long as the infected cell continues to divide.
Special sequences contained within the RNA are duplicated during the reverse tran-scription process so that the integrated provirus contains identical long terminal repeats (LTRs) at its ends. The LTR sequences contain the appropriate promoter, enhancer, and other signals required for transcription of the viral genes by the host RNA polymerase II. Transcription produces a full-length RNA genome and one or more spliced mRNAs. For the oncoviruses, the predominant spliced mRNA is translated to produce the envelope glycoproteins, but in HIV-1, a series of spliced mRNAs are produced that encode, in addi-tion to the envelope proteins, a series of viral regulatory and accessory proteins. Unlike most retroviruses, HIV-1 and the other lentiviruses apparently exert considerable control over whether the primary transcripts are allocated to full-length RNA or are spliced to produce mRNAs . With the exception of these regulatory and accessory pro-teins, all retroviral proteins are initially translated as polyproteins that are subsequently processed by proteolysis into the individual protein molecules. The enzyme responsible for most of these protein cleavages is the virus-specific protease (PR) that is encoded in either the gag gene or the pol gene, depending on the virus (see Table 42 – 1).

A simplified view of retroviral RNA replication is presented in Figure 42 – 3. In addition to DNA polymerase activity, the reverse transcriptase possesses an RNase H activity that is responsible for degrading the RNA portion of the DNA – RNA hybrid ( RNA/ DNA) produced in the first phase of reverse transcription. The immediate product of reverse transcription is a linear double-stranded DNA molecule that is flanked by the LTR se-quences. The viral integrase (IN) catalyzes the integration of the linear DNA into host DNA. The integration process is highly specific with respect to the viral DNA, and two base pairs are generally lost from each end of the DNA. The choice of a target site for in-tegration into the cellular DNA appears, however, to be nearly random. A short sequence of base pairs in the target DNA (four to six, depending on the virus) is duplicated during the integration process, and these repeat sequences immediately flank the integrated provirus. The replication process is completed by transcription of the proviral DNA by the host RNA polymerase II.
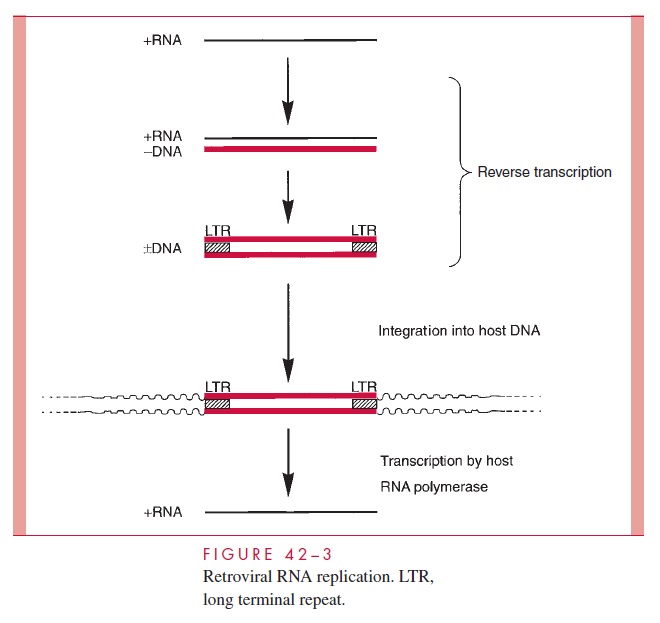
It should be noted that the scheme represented in Figure 42 – 3 also describes the replication cycle for hepatitis B virus . Instead of packaging the RNA form of the genome as occurs with retroviruses, hepatitis B virus packages the double-stranded DNA that is the immediate product of reverse transcription.
Of all the known retroviruses, HIV-1 possesses the most error-prone reverse transcrip-tase. The consequence of this high error rate is that each time the viral RNA is reverse transcribed, one to two new mutations are introduced into the resulting DNA.
Because the process of transcription of the integrated proviral DNA to produce new viral genomes is also error prone, mutant genomes accumulate rapidly over the course of an infection. The end result is a quasispecies that accounts for the many nucleotide differences observed between different isolates (even from the same infected individual) and for the variability of the SU envelope protein gp120. It may explain, in part, the failure of the immune sys-tem to control the infection and also the increases in viral virulence that appear to occur during the course of the infection.
RETROVIRAL GENES
The organization of the genome of different types of retroviruses is shown in Figure 42 – 4 (see also Table 42 – 1). The order of the genes for a typical retrovirus is gag – pol – env. The gag (group-specific antigen) gene encodes the structural proteins of the virus and, in somecases, the protease. The pol (polymerase) gene encodes the reverse transcriptase, the inte-grase, and sometimes the protease. The env (envelope) gene encodes the two membrane glycoproteins found in the viral envelope. Not surprisingly, the SU protein (gp120 in HIV-1) is responsible for the host range of the virus and its antigenicity.
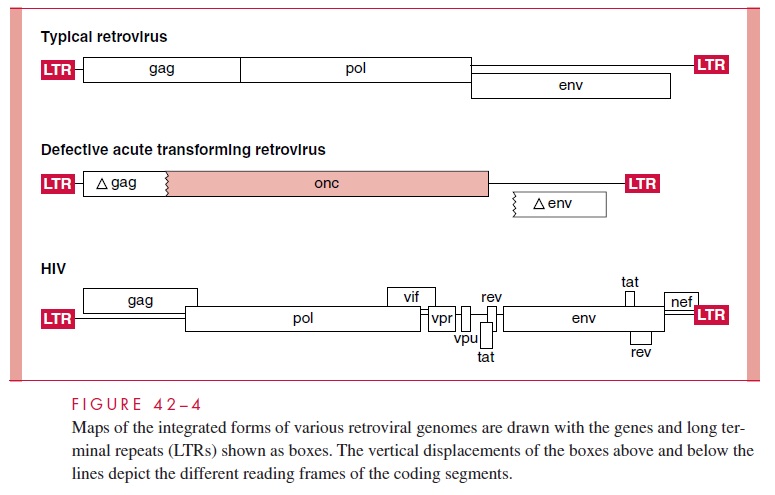
The genomes of acute transforming oncoviruses have a variety of structures, but one feature is common to nearly all of them: some viral genes are replaced by genes derived from their hosts that render them oncogenic . In every case, the signals re-quired for reverse transcription and for transcription of the provirus, which are located near the ends of the RNA, are retained in the infecting virus. In the example shown in Figure 42 – 4, the pol gene and parts of both the viral gag and env genes are deleted, but other configurations are possible. Such oncoviruses are defective and replicate only in the presence of a helper virus that can supply the missing functions.
A comparison of the genetic makeup of HIV-1 with that of a typical retrovirus (see Fig 42 – 4) reveals a larger number of genes and a much more complex organization. HIV-1 contains, in addition to the gag, pol, and env genes, an array of other genes (tat, rev, nef,vif, vpr, and vpu). Expression of these genes requires mRNA splicing, and all apparentlyencode proteins that serve regulatory or accessory roles during the infection . HTLV-1 encodes the regulatory proteins, Tax and Rex, which are analogous to the HIV-1 Tat and Rev proteins.

The names of the genes that have been best characterized and the proteins and functions they determine are listed in Table 42 – 2.
TRANSFORMATION BY RETROVIRUSES
Oncogenic retroviruses appear to transform cells to an oncogenic state by three distinct mechanisms .
First, the defective acute transforming viruses (see Fig 42 – 4) have acquired a cellu-lar gene (thereafter called an oncogene) that when expressed in the infected cell results in loss of normal growth control. On infection, the transduced oncogene is expressed from the viral LTR promoter, resulting in a rapid and acute onset of malignant disease. Persistent transformation by oncogene transduction is possible only for those retro-viruses that are not cytocidal. More than 30 different oncogenes have been identified in a variety of animal retroviruses, but no human retroviruses are known that transform by this mechanism.
The second mechanism is called insertional mutagenesis. Integration of a retro-virus in the vicinity of particular cellular genes can cause inappropriate expression of the gene, resulting in uncontrolled cell growth. These cellular genes are called protooncogenes, and insertional activation by the virus is apparently due to the closeproximity of the integrated viral promoter or enhancer to the gene. Cancers that are caused by this mechanism have very long latent periods, because integration is random and only rarely occurs near a cellular protooncogene.
The causative agent of adult T-cell leukemia, HTLV-1, exemplifies the third mecha-nism. In this case, the integrated provirus in the leukemic cells from any one patient is found at a unique location on a particular chromosome. Thus, the tumors are probably monoclonal. The cancer is not the result of insertional activation, however, because the chromosomal location of the provirus is never the same in any two patients. Instead, transformation results from the continual expression of the viral tax gene (the HTLV-1 homolog of the HIV-1 tat gene; see Table 42 – 2). Apparently, the Tax protein not only can transactivate viral transcription in the same manner as Tat , but Tax can also transactivate the expression of one or more cellular genes (possibly protooncogenes),resulting in malignant transformation.
ROLES OF HIV-1 REGULATORY AND ACCESSORY PROTEINS
A unique feature of HIV-1 and other members of the lentivirus subfamily is the ability to produce a complex array of regulatory and accessory proteins that appear to be responsible for staging the infection, increasing the efficiency and yield of the infec-tion, and in some cases contributing to viral latency. These proteins also appear to in-teract with cellular factors to modulate the infection differently in different host cells. The roles of the two HIV-1 regulatory genes, tat and rev, and the four accessory pro-teins, nef, vpu, vpr, and vif, are discussed below and summarized in Table 42 – 2. Although the four accessory proteins are dispensable in many cell culture systems, they appear to be important for the maximum pathogenic potential of the virus in infected individuals.
The products of the tat and rev regulatory genes are the Tat and Rev proteins, respec-tively. Both of these proteins have the effect of staging the infection, so that in the ab-sence of abundant transcription of the proviral genome, only limited gene expression is possible. When the infected T-lymphocyte is stimulated, for example by antigen presenta-tion, Tat and Rev play a positive role in promoting viral gene expression. In the absence of high levels of Tat, the host RNA polymerase initiates properly at the LTR promoter, but transcription is usually prematurely terminated leading to the production of short, dead-end transcripts. Tat is a transcriptional activator that acts at a sequence near the beginning of the viral mRNA, called TAR, to recruit cellular proteins to the transcribing RNA poly-merase, resulting in a modification to the polymerase that prevents premature termination and allows complete transcription of the proviral genome.
The Rev protein acts at the level of mRNA splicing. Normally, unspliced cellular tran-scripts are retained in the nucleus and only fully spliced mRNAs are transported to the cytoplasm for translation. The only viral proteins that are made from fully spliced mRNAs are Tat, Rev, and Nef, and consequently only these proteins are found early after infection, when there is no mechanism to prevent complete splicing of pre-mRNAs. To express the Vif, Vpr, and Vpu proteins, and the Env polyprotein, which are all made from singly spliced transcripts, as well as the Gag and Pol polyproteins, which are translated from the unspliced genomic RNA, it is necessary to transport incompletely spliced RNAs to the cytoplasm. Transport of partially spliced transcripts is accomplished by Rev bind-ing to a site on the viral RNA within the env gene called the Rev-responsive element (RRE). The RNA-bound Rev then interacts with normal cellular machinery responsible for protein export from the nucleus to mediate the movement of the RNA through the nu-clear pore. By promoting translation of the virion structural proteins and some of the accessory proteins, Rev turns up late gene expression that leads directly to a high rate of virus production.
The Nef accessory protein enhances virus production and virion infectivity and also ap-pears to interfere with immune recognition of infected cells. Nef causes the internalization and degradation of the CD4 protein, which likely contributes to virus release by preventing the formation of complexes between the cellular receptor and newly synthesized virions. Nef also causes the downregulation of cell surface major histocompatibility com-plex (MHC) I molecules, which may prevent recognition of infected cells by cytotoxic T lymphocytes. In addition, virions produced in the absence of the Nef protein are at least partially blocked at some step prior to integration. The combination of these and perhaps other effects allows the Nef protein to play an essential pathogenic role in an infected in-dividual.
The Vpu protein appears to play two separate roles during the late stages of infection. In the absence of Vpu, the Env protein forms complexes with CD4 in the endoplasmic reticulum and fails to reach the plasma membrane of the cell. One of the roles of Vpu is to target the destruction of CD4 in the endoplasmic reticulum to allow for incorporation of Env into newly synthesized virions. The second role of Vpu is to promote the release of virions from the infected cell by an unknown mechanism.
The Vpr protein is involved in promoting import of subviral particles into the nucleus after reverse transcription. Thus, the protein has little or no effect in proliferating T cells where nuclear access is ensured with each mitosis. However, successful infection of non-dividing cells such as macrophages requires Vpr to allow the newly synthesized viral DNA to reach the nucleus and be integrated into the cellular DNA.
Vif (virion infectivity factor) increases the infectivity of HIV-1 in primary T cells and certain “nonpermissive” cells in culture. In the absence of Vif, the virus fails to complete reverse transcription in these cell types. “Permissive” cell lines infected by mutants defec-tive in the vif gene produce normal yields of infectious virus. One possible explanation for this observation is that “permissive” cells contain a factor that can substitute for the missing Vif protein. Thus, one role of Vif may be to extend the host range of HIV-1 to cell types that would otherwise not be infected.
Superimposed on this complex regulatory network is the fact that the viral pro-moter contains elements that are sensitive to specific cellular transcription factors. This observation may help explain why virus production in CD4 T lymphocytes is greatly increased when the cells are activated. Clearly the outcome of an HIV-1 infec-tion is determined by a complex interplay between a very large number of different factors.
Related Topics