Chapter: Modern Medical Toxicology: Analytical Toxicology: Analytical Instrumentation
Quantitative Assays - Analytical Toxicology Instrumentation
QUANTITATIVE ASSAYS
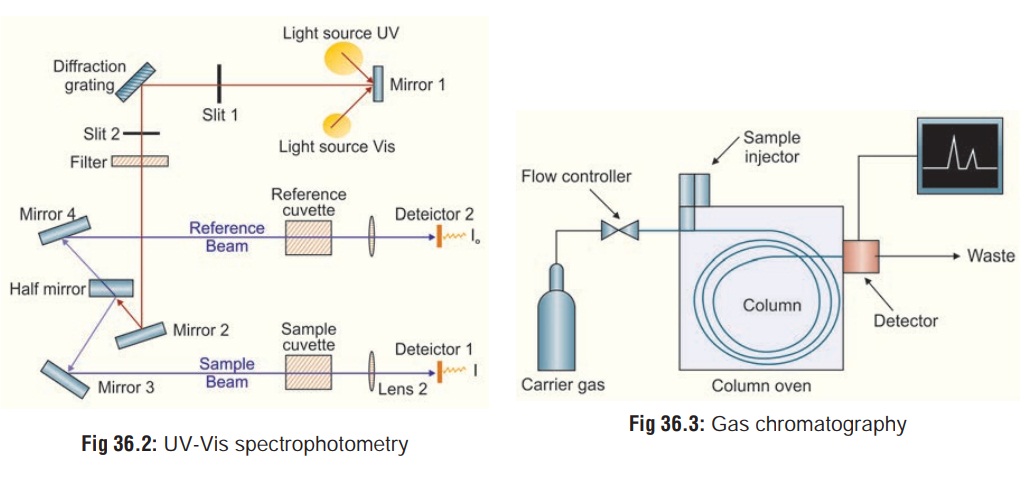
1. Ultraviolet-Visible (UV-Vis) Spectrophotometry
This
technique is based on the principle that many drugs when in solution will
absorb UV radiation. The degree of absorption depends on the chemical structure
of the drug, its
concentration in the solution, and the wavelength of the UVR. The functioning
of this instrument is relatively straightforward. A beam of light from a
visible and/or UV light source is separated into its component wavelengths by a
prism or diffraction grating. The monochromator can select from the light
source an ultraviolet ray of any given wavelength ranging from 200 to 340 nm.
Thesample extract in aqueous medium is placed in a transparent quartz cuvette
in the path of the radiation. Each monochromatic (single wavelength) beam in
turn is split into two equal inten-sity beams by a half-mirrored device. One
beam, the sample beam passes through a small transparent container (cuvette)
containing a solution of the compound being studied in a trans-parent solvent.
The other beam, the reference, passes through an identical cuvette containing
only the solvent. The intensities of these light beams are then measured by
electronic detectors and compared (Fig
36.2). The intensity of the reference beam, which should have suffered
little or no light absorption, is defined as I0. The intensity of the sample beam is defined as I. Over a short period of time, the
spectrometer automaticallyscans all the component wavelengths in the manner
described. The ultraviolet (UV) region scanned is normally from 200 to 400 nm,
and the visible portion is from 400 to 800 nm.
If the sample compound does not absorb light of a given wavelength, I = I0. However, if the sample compound absorbs light then I is less than I0, and this difference may be plotted on a graph versus wavelength. Absorption may be presented as transmittance (T = I/I0) or absorbance (A = log I0/I). If no absorption has occurred, T = 1.0 and A = 0. Most spectrometers display absorbance on the vertical axis, and the commonly observed range is from 0 (100% transmittance) to 2 (1% transmittance). The wavelength of maximum absorbance is a characteristic value, designated as λmax. Different compounds may have very different absorption maxima and absorbances. Intensely absorbing compounds must be examined in dilute solution, so that significant light energy is received by the detector, and this requires the use of completely transparent (non-absorbing) solvents. The most commonly used solvents are water, ethanol, hexane and cyclohexane. Solvents having double or triple bonds, or heavy atoms (e.g. S, Br & I) are generally avoided. Because the absorbance of a sample will be proportional to its molar concentration in the sample cuvette, a corrected absorption value known as the molar absorptivity is used when comparing the spectra of different compounds. Molar absoptivities may be very large for strongly absorbing compounds (ε >10,000), and very small if absorption is weak (ε = 10 to 100).
The amount of UV radiation which
passes through the solution is measured by the photocell. By steady rotation of
the monochromator, it is possible to pass sequentially UV rays of all
wavelength from 200 to 340 nm through the extract. The photocell monitors the
amount of radiation absorbed at each wavelength and this is transcribed on to a
recorder chart. The shape of the spectrum, the wavelength at which absorption
is at a maximum, and any changes brought about by changing the pH of the
extract are all used as an aid to characterise the agent present. This
technique is ideal to quantitate blood levels of paracetamol and salicylates,
as well as urine levels of phenothiazines.![]()
A major disadvantage of UVS is the
possibility of interfer-ence in multiple drug overdose. In such a case, UV
scanning can produce a composite spectrum of bewildering complexity from which
neither qualitative nor quantitative information can be derived. Conventional
spectrophotometric methods are known for producing false positive
results which can be disastrous in medico-legal cases.
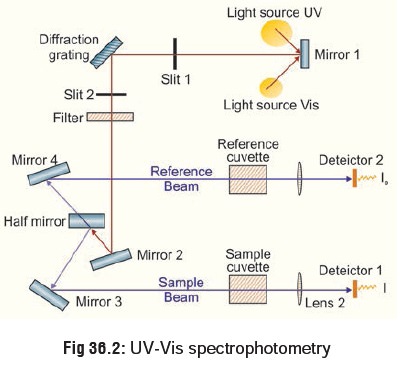
2. Gas Chromatography (GC)
This is a more sophisticated system of
quantitative analysis and has found great favour with analytical toxicologists
since it offers a way of simultaneously separating, identifying, and measuring
drugs and other organic poisons. Gas chromatog-raphy, specifically gas-liquid
chromatography, involves a sample being vapourised and injected onto the head
of the chromato-graphic column. The sample is transported through the column by
the flow of inert, gaseous mobile phase. The column itself contains a liquid
stationary phase which is adsorbed onto the surface of an inert solid. The
liquid or solid specimen dissolved in a solvent is injected into the
chromatograph. The specimen is vapourised by heat and is carried through a
column by an inert carrier gas (usually nitrogen). The choice of carrier gas is
often dependent upon the type of detector which is used. The carrier gas system
also contains a molecular sieve to remove water and other impurities. The
column is packed with a substance like carbowax or adiponitrile which is
capable of changing the migration time of the specimen as it traverses the
column.
For optimum column efficiency, the
sample should not be too large, and should be introduced onto the column as a
“plug” of vapour - slow injection of large samples causes band broad-ening and
loss of resolution. The most common injection method is where a microsyringe is
used to inject sample through a rubber septum into a flash vapouriser port at
the head of the column. The temperature of the sample port is usually about
500C higher than the boiling point of the least volatile component of the
sample. For packed columns, sample size ranges from tenths of a microlitre up
to 20 microlitres. Capillary columns, on the other hand, need much less sample,
typically around 10-3 microlitre. For capillary GC, split/splitless injection
is used.
The injector can be used in one of
two modes; split or splitless. The injector contains a heated chamber containing
a glass liner into which the sample is injected through the septum. The carrier
gas enters the chamber and can leave by three routes (when the injector is in
split mode). The sample vapourises to form a mixture of carrier gas, vapourised
solvent![]() and vapourised solutes. A proportion
of this mixture passes onto the column, but most exits through the split
outlet. The septum purge outlet prevents septum bleed components from entering
the column.
and vapourised solutes. A proportion
of this mixture passes onto the column, but most exits through the split
outlet. The septum purge outlet prevents septum bleed components from entering
the column.
A detector recognises the presence
of the chemical and graphically plots its emergence as a function of time (Fig 36.3). The retention time and peak
area for a chemical compared to known standard are used to identify and to
quantitate its presence. There are many detectors which can be used in gas
chromatography. Different detectors will give different types of selectivity. A
non-selective detector responds to
all compounds except the carrier gas, a selective
detector responds to a range of compounds with a common physical or
chemical property and a specific detector
responds to a single chemical compound. Detectors can also be grouped into concentration dependentdetectors and mass flow dependent detectors. The
signal froma concentration dependant detector is related to the concentra-tion
of solute in the detector, and does not usually destroy the sample. dilution
with make-up gas will lower the detectors response. Mass flow dependant
detectors usually destroy the sample, and the signal is related to the rate at
which solute molecules enter the detector. The response of a mass flow
dependant detector is unaffected by make-up gas.
The most common means of detecting
the compounds is by flame ionisation. On leaving the column the inert gas is
mixed with hydrogen and either air or oxygen, and the mixture is ignited to give
a continuous jet of flame. It is decomposed into electrically charged fragments
which are collected at an electrode. This results in an electrical signal which
is amplified and transmitted to a pen recorder operating at a constant chart
speed. This peak is transcribed each time a compound emerges from the column.
Flame ionisation detectors (FIDs) are mass sensitive rather than concentration
sensitive; this gives the advantage that changes in mobile phase flow rate do
not affect the detector’s response. The FID is a useful general detector for
the analysis of organic compounds; it has high sensitivity, a large linear
response range, and low noise. It is also robust and easy to use, but
unfortunately, it destroys the sample.
Gas chromatography (GC) is most
commonly employed to quantitate blood levels of volatile liquids such as
ethanol, ethylene glycol, and methanol.
Other detectors have recently been
developed which widen the scope of plasma screening, such as the electron capturedetector, and the alkali flame detector (nitrogen detector).
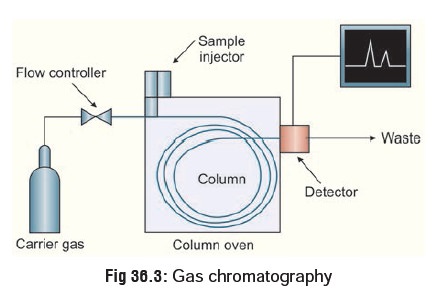
High Performance Liquid Chromatography (HPLC)
This is similar to GC, except that
it is not restricted to volatile compounds. A high pressure (1000 to 6000 pound
per square inch) pump facilitates movement of the specimen through the columns
packed with chromatographic adsorbents e.g. silica gel and alumina. The
effluent stream passes through a detector, usually an ultraviolet
spectrophotometer, and the appearance of a drug in the solvent is signalled by
a recorder peak in the same way as in GC (Fig
36.4). Again, the size of the peak is proportional to the
concentration of drug in the sample. HPLC can be used to separate and analyse
complex mixtures.
Related Topics