Chapter: Pharmaceutical Biotechnology: Fundamentals and Applications : Recombinant Human Deoxyribonuclease I
Protein Chemistry, Enzymology, and Structure
PROTEIN CHEMISTRY, ENZYMOLOGY, AND STRUCTURE
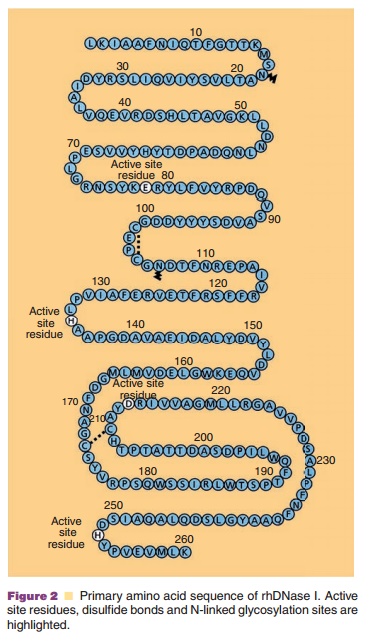
The protein chemistry of human DNases including DNase I, has been recently reviewed by Lazarus (2002) and Baranovskii et al. (2004). Recombinant human DNase I is a monomeric, 260-amino acid glycoprotein (Fig. 2) produced by mammalian CHO cells (Shak et al. 1990). The protein has four cysteines, which are oxidized into two disulfides between Cys101-Cys104 and Cys173-Cys209 as well as two potential N-linked glycosylation sites at Asn18 and Asn106 (Fig. 2). rhDNase I is glycosylated at both sites and migrates as a broad band on polyacrylamide gel residues (Glu78 and Asp 212), all of which are critical for the general acid–base catalysis of phosphodiester bonds since alanine substitution of any of these results in a total loss of activity (Ulmer et al., 1996). The two Ca2þ binding sites require acidic or polar residues for coordination of Ca2þ; for site 1 these include Asp201 and Thr203 and for site 2 these include Asp99, Asp107, and Glu112 (Pan and Lazarus, 1999). Other residues involved in the coordination of divalent metal ions at the active site and DNA contact residues have been identified by mutational analysis (Pan et al., 1998a). DNase I is a relatively stable enzyme and shows optimal activity at pH 5.5 to 7.5. It is inactivated by heat and is potently inhibited by EDTA and G-actin. Surprisingly, DNase I is also inhibited by NaCl and has only ca. 30% of the maximal activity in physiological saline.
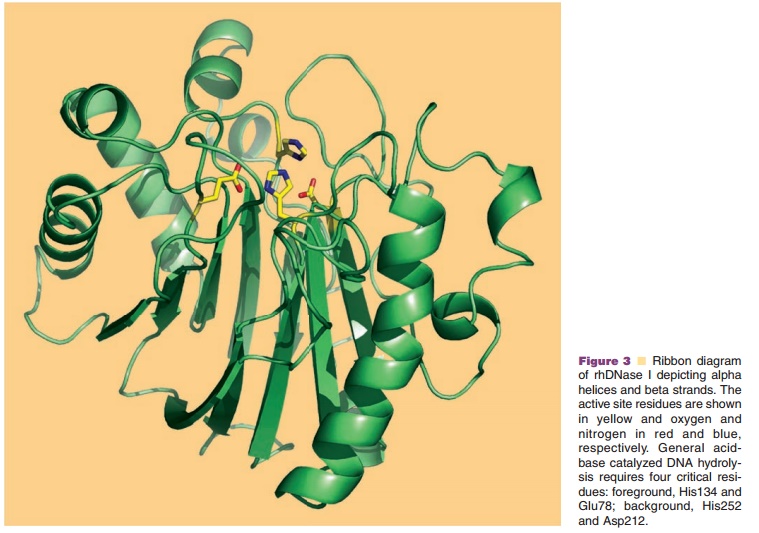
The X-ray crystal structure of rhDNase I has been solved at 2.2 A resolution and superimposes with the biochemically more widely studied bovine DNase I, which shares 78% sequence identity, with an rms deviation for main chain atoms of 0.56 A (Wolf et al., 1995). DNase I is a compact α/β protein having a core of two tightly packed six-stranded β-sheets surrounded by eight α-helices and several loop properties have been engineered by site-directed mutagenesis. The methods for produc-tion of the variants and the assays to characterize them have been reviewed recently (Pan and Lazarus, 1997; Sinicropi et al., 2001). The rationale for improving activity was to increase binding affinity to DNA by introducing positively charged residues (Arg or Lys) on rhDNase I loops at the DNA binding interface to form a salt bridge with phosphates on the DNA backbone. These so-called “hyperactive” rhDNase I variants are substantially more active than wildtype rhDNase I, and are no longer inhibited by physiological saline. The greater catalytic activity of the hyperactive variants is due to a change in the catalytic mechanism from a “single nicking” activity in the case of wildtype rhDNase I to a “processive nicking” activity in the hyperactive rhDNase I variants (Pan and Lazarus, 1997), where gaps rather than nicks result in a higher frequency of double strand cleavages.
It is interesting to note that significantly greater activity can result from just a few mutations on the surface that are not important for structural integrity. For whatever reason DNase I is not as efficient an enzyme as it could be for degrading DNA into small fragments. Furthermore the inhibition by G-actin can be eliminated by a single amino acid substitution (see below). Thus DNase I is under some degree of regulation in vivo. One can only speculate that nature may have wanted to avoid an enzyme with too much DNA degrading activity that could result in unde-sired mutations in the genome.
Related Topics