Chapter: Biochemical Pharmacology : Pharmacodynamics
Partial agonism and the two-state model of receptor activation
Partial agonism and the
two-state model of receptor activation
The efficacy will obviously
vary for drugs that act on the same physiological parameter by different
routes; e.g., morphine is a stronger painkiller than aspirin is. Howev-er,
profound differences may even be observed with sub-stances that act on the very
same site of the very same tar-get molecule. Figure 3.11 shows an example. The
recep-tor in question is a serotonin receptor (subtype 1A) which occurs in the
brain and is the target of some psychoactive drugs. Like the adrenergic
receptors mentioned above, it is a G protein-coupled receptor. Receptor activation
will trig-ger exchange of GDP for GTP in the cognate G proteins. It can
therefore be measured by way of incorporation of GTP-γ35S, which is both radioactive and resistant to the in-trinsic
GTP'ase activity of the G protein. You can see that the effects of the
different agonists applied not only arise at different concentrations but also
level off at different max-ima, some of them well below the reference value
(100%).
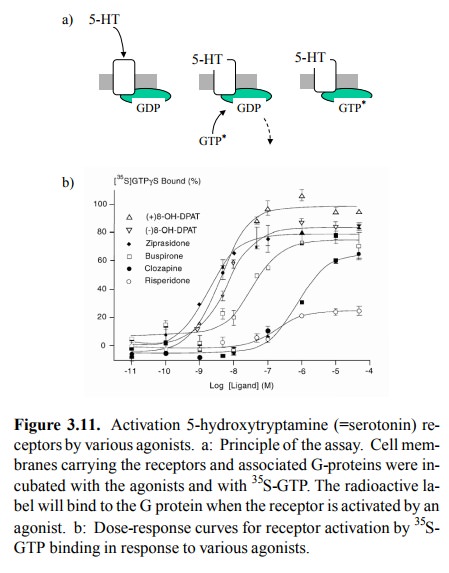
Agonists that show
sub-maximal activation of a receptor, even at saturating concentrations, are
called `partial ago-nists'. How can an agonist be `partial'?
A plausible explanation can
be given if we consider the al-losteric nature of the `typical' receptor
protein and its interaction with the ligand7. An allosteric protein
has two con-formational states that are in equilibrium (Figure 3.12). With most
receptors, the inactive state will be more preva-lent in the absence of
agonists. However, an agonist will exclusively bind to and therefore favour the
active confor mation. If the concentration of agonist is sufficiently high, the
entire receptor population will be arrested in the ac-tive state.
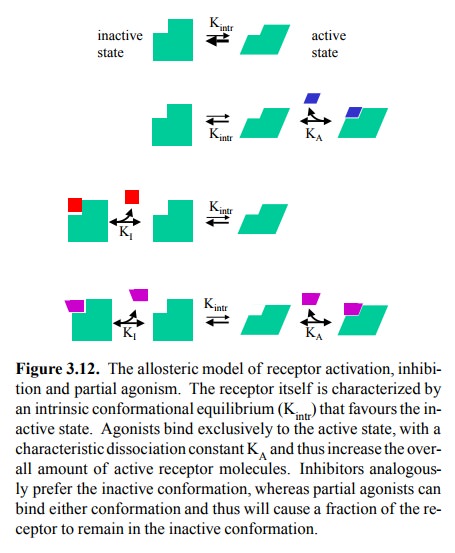
Analogously, an antagonist
will preferentially bind the in-active conformation and therefore, at
saturating concentra-tions, convert the entire receptor population to the
inactive state. A partial agonist
will have finite affinity for both con-formational states of the receptor,
although it will be higher for the active conformation, so as to overcome the
intrinsic preference of the inactive state (Kintr in Figure 3.12)
and therefore bring about any appreciable receptor activation at all.
Differences in the maximum effect (or efficacy) be-tween different partial
agonists then simply correspond to different ratios of KA/KI,
as defined in Figure 3.12.
As you can see in Figure 3.12
(bottom row), the receptor assumes four distinct states:
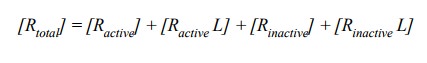
The active fraction of the
receptor comprises the free and the ligand-associated active forms:
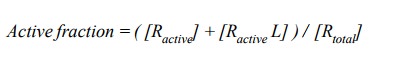
The expression that that
quantitatively describes the active fraction as a function of the ligand
concentrationcan quite easily be derived using the equilibrium equations,
together with the two equations above:

I will post an Excel
spreadsheet on the course website that will allow you to play a bit with the
parameters and see how they affect the resulting curve.
Are partial agonists clinically
relevant? Once upon a time, partial agonists for β-adrenergic receptors were promot-ed as a
`milder' alternative to β-blockers, but this seems to have been
abandoned. A practically more interesting appli-cation are synthetic opioids.
These are being used for anal-gesia, i.e. as pain killers. While morphine or
other full ag-onists such as fentanyl are the strongest pain killers
avail-able, partial agonists such as nalorphine or pentazocine are being used
in applications requiring something interme-diate in strength between morphine
and basic pain killers such as aspirin. The rationale for preferring partial
agonists over the `real thing' is that the partial agonists also seem to have a
lower addictive effect than the full agonists, al-though this is not undisputed.
Related Topics