Chapter: Biochemical Pharmacology : Pharmacodynamics
Mechanisms and kinetics of drug receptor interaction
Mechanisms and kinetics of
drug receptor interaction
There are several typical
mechanisms of action that apply to the different types of receptor proteins.
For enzymes, these are
• Competitive inhibition: The drug occupies the
active site and prevents binding of the physiological substrate. Example: The
inhibition of angiotensin convertase by enalapril.
• Irreversible (covalent) inhibition: The drug
again binds to the active site of the enzyme and then covalently reacts with
it, so that the active site becomes irreversibly blocked. Example: Inhibition
of cyclooxygenase by acetylsalicylic acid.
• Allosteric inhibition: The drug binds outside
the active
site but prevents the enzyme
from adopting its active conformation. Example: Inhibition of Na+/K+-ATP'ase
by digitoxin or digoxin.
The allosteric behaviour seen
with many enzymes is also typically observed with ion channels and metabolic
recep-tors. In the absence of physiological agonists, these proteins typically
prefer their inactive conformation; channels will be closed, and metabolic
receptors will not stimulate their downstream cascades. The physiological
agonists act al-losteric activators, promoting conversion to the active state.
Drugs acting on these targets typically belong to one of the following classes:
• Reversible agonists (activators), i.e. the drug mimics the physiological agonist.
Example: Isoproterenol, an agonist at β-adrenergic receptors.
• Reversible inhibitors: The drug, typically in a
competitive way, prevents binding of the physiological agonist. Example:
Propranolol, an antagonist at β-adrenergic re-ceptors.
• Reversible partial agonists: The drug has
activity intermediate between that of an inhibitor and an agonist. Ex-ample:
Dobutamine, a partial agonist at β-adrenergic re-ceptors. Partial agonists may be
used for their agonistic properties or their antagonistic properties.
• Irreversible (covalent) inhibitors. This case
is less common than reversible inhibition or activation. Example:
Phenoxybenzamine, an antagonist at α-adrenergic re-ceptors.
With few exeptions, all drugs
we are going to consider in the rest of this course will fall into one of the
above cat-egories.
1. Mass action kinetics of drug-receptor binding
In the simplest possible
case, one effector molecule, which may be either the physiological agonist or a
drug, will bind to one target molecule, and all target molecules will bind the
effector with the same affinity. It is noteworthy that there are numerous
deviations from this simple situation1. Nevertheless, we will
confine ourselves to this simple mod-el, which will still take us to some
important conclusions.
With the above assumptions,
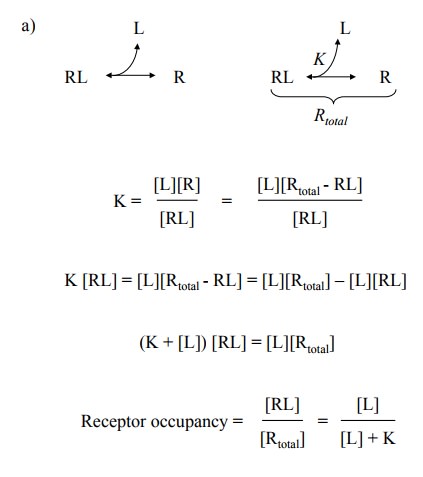
the binding will be subject to the law of mass action, and a single parameter –
the disso-ciation constant, typically called K – will describe the
inter-action. K will be an empirical value, depending on both the ligand and
the receptor molecule in question. K is inversely related to the affinity; the
higher it is, the lower the binding affinity. The law of mass action can be
rearranged to give us the receptor occupancy, i.e. the fraction of all
receptors saturated with the ligand (Figure 3.1a). You will recognize the
formal similarity to Michaelis-Menten enzyme kinetics. Accordingly, if we plot
the receptor occupancy as a func-tion of the ligand concentration, we get the
same hyperbolic type of curve (Figure 3.1b, top).
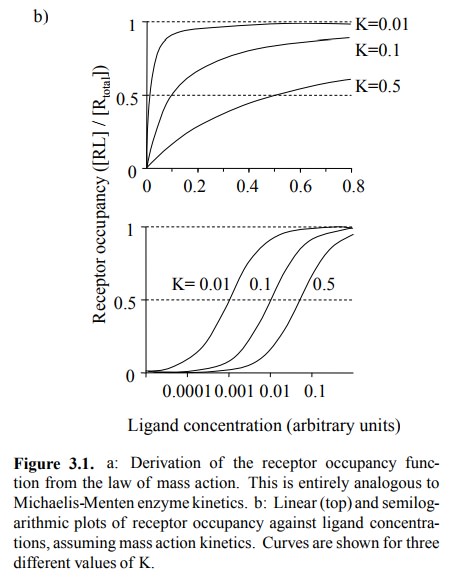
Shown are three curves,
differing in their respective values for K. The bottom panel shows that
plotting the same num-bers on a logarithmic scale for the ligand yields nice
sig-moidal plots, which are now distinguished solely by their parallel offsets
along the x-axis. From these plots, K can be determined as the ligand
concentration of half-maximal receptor occupancy.
If a drug activates its receptor, it simply
assumes the role of the ligand in the above model, albeit its affinity will
most likely differ from that of the physiological ligand. What we can see,
then, is that very little benefit can be expected from increasing the drug
concentration beyond, say, five times its K value, since the receptor will
already be saturated. The only thing that will happen upon further increase is
that secondary, less affine and specific sites will be bound, potentially
evoking unwanted side effects.
If the drug is an inhibitor, we are dealing with a ternary
sys-tem of receptor, physiological agonist, and our inhibitory drug. We will
examine two cases: Reversible competitive inhibitors (Fig. 3.2, top) and
irreversible ones (Fig. 3.2, bottom).
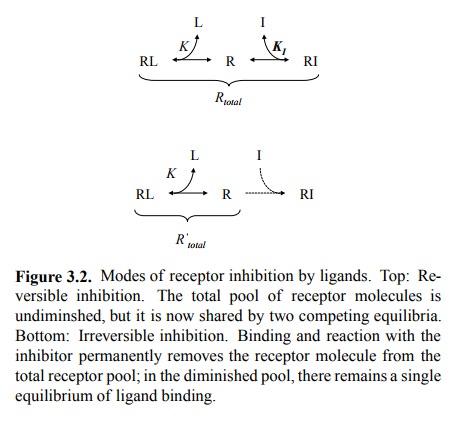
2. Reversible inhibition
If a drug does not undergo a
covalent reaction with its re-ceptor, binding will almost always be reversible2.
There fore, the total number of functional receptor molecules will not change,
but we now have two linked, competing equi-libria squeezed into the same pool.
This gives rise to a mod-ified relationship of receptor occupancy to ligand
concen-tration, as stated and illustrated in Figure 3.3. Again, the situation
is entirely analogous to reversible inhibition in Michaelis-Menten kinetics3,
and you may want to consult your biochemistry textbook for the derivation – or
just do it yourself, as an exercise.
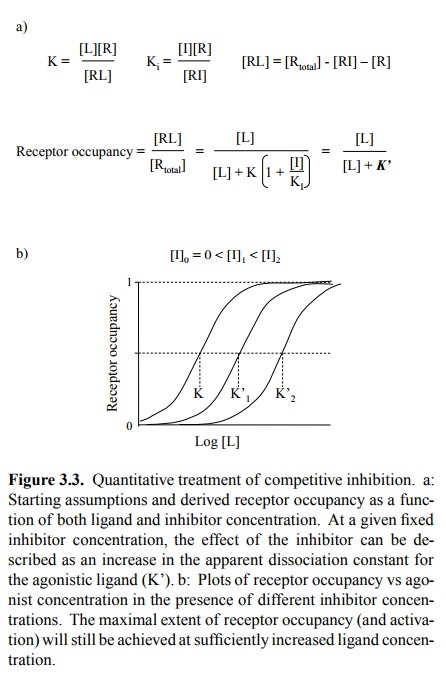
An important aspect of competitive
inhibition is that, with sufficiently high concentrations of physiological
ligand, the receptor can still be maximally activated. Competitive inhibition
thus reduces the receptor's sensitivity to the ago-nist but does not diminish
the maximum effect that can be attained at very high agonist concentrations.
This means that, in case of an accidental overdose of the inhibitor, the
endogenous agonist or a drug that mimics it could be used to overcome the
inhibition.
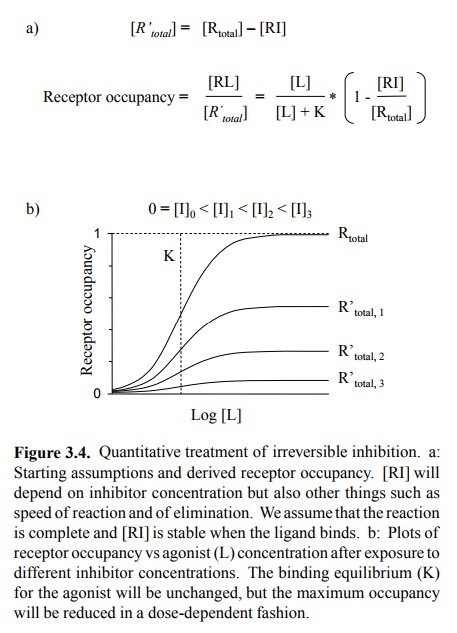
3. Irreversible inhibition
If a drug undergoes a
covalent reaction with its receptor, the receptor molecules affected will be
irreversibly blocked and thus altogether removed from the total receptor pool
available for the interaction with the agonist. Thus, the agonist-receptor
equilibrium now plays out in that reduced total pool. The number of occupied
receptors will therefore be proportionally reduced (Figure 3.4).
4. Example: Inhibition of α-adrenergic receptors by
tolazoline and phenoxybenzamine
For an experimental
illustration of the foregoing, let us look at the inhibition of α-adrenergic receptors. These re-ceptors are stimulated by
epinephrine and norepinephrine; stimulation will increase the tension of blood
vessel walls and therefore enhance blood pressure. α-Adrenergic re-ceptors are very numerous in the spleen. The spleen
has a sponge-like structure and stores about half a litre of blood, which upon
adrenergic stimulation will get squeezed out into the circulation4.
This extrusion of blood is effected by the contraction of smooth muscle cells
that are embedded in the spleen tissue. Accordingly, if we take a fresh slice
of spleen and bathe it in solutions of mediators or drugs, we can measure its
mechanical tension to quantify the extent of α-adrenergic stimulation. Figure 3.5a shows the
force of contraction developed by such spleen strips in response to varying
concentrations of norepinephrine, in the presence of tolazoline or
phenoxybenzamine, respectively. By com parison to the theoretical plots above
(Fig. 3.3, 3.4), you will be able to decide which of the two inhibitors is the
re-versible one, and which is the covalent one.
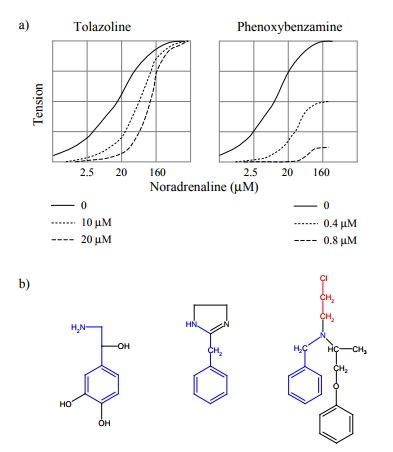
Let us consider the molecular
principles behind the two modes of inhibition. Fig. 3.5b shows the structures
of the agonist (norepinephrine) and of the two inhibitors. With some
imagination, one can spot the similarity between agonist and inhibitors, so
that it is understandable that they all bind to the same site on the α-adrenoceptor. Tolazoline has no obvious reactive groups, and it
will therefore bind non-covalently and reversibly.
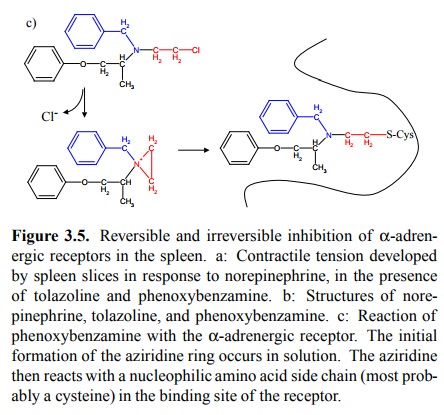
Phenoxybenzamine, on the
other hand, has a chloroethyl group (indicated in red) attached to the nitrogen
that is quite reactive. It will undergo the reactions depicted in Figure 3.5c.
The initial step results in the formation of an ethylen-imine group, which is
quite reactive because of the ring tension. In a second step, after binding to
the receptor, the ring is opened by some nucleophile, most probably the SH
group of a cysteine5 that is part of the receptor molecule. In this
way, the drug becomes covalently attached to the recep-tor and permanently
inactivates it.
Several things are notable
about the action of phenoxyben-zamine:
• The initial circularization (formation of the
aziridine ring) is rather slow, causing the pharmacological action to lag
behind the plasma levels. On the other hand, re-ceptor blockade will persist
long after any excess drug has been eliminated. With most drugs that act by
non-covalent association with their receptors, plasma levels correlate much
more closely with the intensity of drug action.
• While the benzylamino moiety of
phenoxybenzamine (blue in Figure 3.5b) targets it to the α-adrenoceptor, the chemical reactivity of the ethyleneimino group
is rather non-selective and will cause molecules not bound to the receptor to
react in random locations, potentially causing harm including genetic damage.
Accordingly, phenoxybenzamine is not the drug of first choice in most clinical
indications of α-adrenoceptor blockade.
Phenoxybenzamine is the drug of choice in one particu-lar
disease called phaeochromocytoma.
This is a tumour of the adrenal glands that produces and intermittently releas-es
very large amounts of epinephrine and norepinephrine, causing dangerous spikes
in blood pressure. The superior effect of phenoxybenzamine in phaeochromocytoma
is a direct consequence of its covalent mode of binding: The inactivated
receptor cannot be reactivated by whatever amounts of hormone released (cf.
Figure 3.5a). In contrast, reversible inhibition could be overridden in this
particular situation.
Related Topics