Chapter: Medicine Study Notes : Musculo-Skeletal
Orthopaedic Tumours
Orthopaedic Tumours
·
Primary bone tumours are rare.
·
Myeloma accounts for half of
malignant bone neoplasms:
o Old, M > F, pathogenic fractures, pepper-pot skull, normocytic
anaemia with Rouleaux
o Gross: red current jelly lesions
·
Classification based on histology
of tumour cell – cell of origin is unknown/debated. Diagnosis difficult.
Requires clinician, radiologist, pathologist
·
Classification:
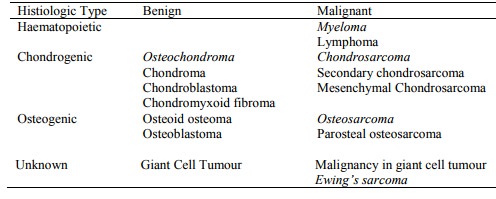
Chondrosarcoma
·
Malignant tumour of cartilage,
with no tumour osteoid or bone being
formed
·
Pain becomes severe and
persistent, swelling
· Typically age 40 – 60 (average 45 years)
·
Most common in the medullary
cavity of the flat bones of the pelvis, large limb bones and ribs. Rare to
involve the extremities
·
Types:
o Conventional: eg diaphysis or metaphysis of long bones. Margins poorly defined. Eroded or
o thickened cortex. X-ray: fluffy
calcification. Grossly, pearly
blue/white colour of cartilage
o Secondary to multiple exostosis in chondrodysplasia
o Dedifferentiated
·
Treatment: tend to metastasise
late (to lung and other bones) ® attempt local excision and replacement with prosthesis
·
Prognosis: Grade 1 and 2 80 – 90%
5-year survival, Grade 3 (rare) 40% 5-year survival. Local or distant
metastasis may occur up to 20 years later
Osteosarcoma (Osteogenic Sarcoma)
·
Proliferating malignant
spindle-cell stroma producing osteoid
·
After multiple myeloma, it is the
most common primary malignant bone tumour
·
50 – 60% of cases are near the
knee (either distal femur or proximal tibia)
·
Types:
o Conventional osteosarcoma: Most common.
Adolescent growth (age 10 – 20). M:F = 2:1, eg
o metaphyses of distal femur and proximal tibia. X-ray: geographic destruction, dense
of lytic, raised periosteum. May also
appear fibroblastic or predominantly chondroid. Gross: haemorrhagic. Micro:
osteoid formation, malignant spinal cells (often see spindle cells in
mesencymal tumours)
o Second, smaller peak in 60 – 70s, secondary to existing disease (eg in
< 1% of Paget‟s), previous irradiation, etc. Microscopically look like a
osteosarcoma
o Also Telangiectatic osteosarcoma, low-grade osteosarcoma, small cell
osteosarcoma (like Ewing‟s but produces osteoid) and surface osteosarcomas (on
the surface of the bone)
·
Investigations: X-ray, serum ALP
(markedly Â), and biopsy
·
Very aggressive: assumed to have
metastasised at diagnosis – usually to lung (in preference to lymph nodes)
·
Treatment: chemotherapy ®
resection ® prosthesis ® post-op adjuvant chemo (high dose methotrexate)
·
5 year survival 60%
Other
·
Benign:
o Osteochondroma: most common benign tumour of bone. Growth of an aberrant focus of cartilage on the surface of the bone (?adherent growth plate). Cartilage-capped lateral bony projection from the metaphysis, usually long bones. Also know as an exostosis. Can be hereditary (® multiple).
o Symptoms due to size, impingement or fracture. X-ray: mushroom like growth from metaphysis. Regular shape. If irregular then ?malignant
o Enchondroma: benign cartilaginous neoplasm usually arising in the
medullary cavity of bone. Most common in age 20 – 50 in small bones of the hand
or foot. Usually clearly circumscribed. Differential is chondrosarcoma –
suspect if large bone in an older patient, erosion of the cortex or suspicious
histology
o Chondroblastoma: benign chondroid neoplasm at the end of long bones
during teens
o Osteogenic tumours: produce osteoid:
§ Osteoid osteomas: Rare. Males in teens. Exquisite pain especially at
night relieved by aspirin. Well-circumscribed lesion of bony trabeculae, with
variable mineralisation. < 1.5 cm. X-ray: radiolucent central zone
surrounded by opaque sclerotic bone
§ Osteoblastoma: Roughly speaking, an osteoid osteoma that is > 1.5 cm
·
Fibrous dysplasia: developmental
defect of bone formation ® enlargement and distortion of the bone. Feels firm, fibrous and may be
gritty
· Malignant:
o Ewing‟s Tumour: Rare. t(11;22)(q24;q12) usual ® fusion gene is an oncogene. Usually age 5 – 10. Usually shaft of long bones, presenting with localised pain or swelling. Small round blue cell tumour. ?neural origin. 75% 5-year survival. Can mimic osteomyelitis
o Giant Cell Tumour: F > M, age 20 – 40, ends of long bones, lytic
lesions, contains multinucleated giant cells. Usually benign. High local
recurrence, rarely metastasises
o Fibrosarcoma
§ Malignant tumour of fibroblasts (ie collagen producing cells)
§ Occurs in any connective tissue but more common in the extremities and
middle aged
§ Fibrosarcoma of the bone is rare.
Swelling, pain, pathological fracture
o Synoviosarcoma:
§ Rare malignant tumour of the synovium, usually sharply circumscribed
§ Rapid enlargement of the joint with pain. Usually knee, hip or shoulder
§ May extend along fascial lines and invade bone
§ Treatment: if small then excise, if high grade: resection + radiotherapy
+ chemotherapy
Secondary Bone Cancer
·
Most common bone cancer Ăž always
ask about previous primaries
·
Source: breast > prostate >
kidney > lung > thyroid
·
Sites: vertebrae, pelvis,
proximal femur, humerus
·
Spread: usually
haematogenous. Occasionally local
extension
·
Usually osteolytic ®
pathological fractures
·
Presentation:
o Pain + history of cancer in 50 – 70 year old
o In children < 6 years: from neuroblastoma
o Symptoms of hypercalcaemia: anorexia, nausea, weakness, depression,
polyuria
·
Investigations:
o Xray: usually osteolytic lesions (if osteoblastic probably carcinoma)
o Bone scan, FBC, ALP, Electrophoresis (myeloma)
o FNA: determining cell of origin helps guide management
·
Treatment: usually palliative,
control pain, prophylactic fixation, spinal stabilisation, radiotherapy (ÂŻ pain)
Related Topics