Chapter: Medical Immunology: Phagocytic Cells
Laboratory Evaluation of Phagocytic Function
LABORATORY EVALUATION OF PHAGOCYTIC FUNCTION
The evaluation of phagocytic function is usually centered on the study of neutrophils, which are considerably easier to isolate than monocytes or macrophages. Phagocytosis by neutrophils can be depressed as a result of reduction in cell numbers or as a result of a func-tional defect. Functional defects affecting every single stage of the phagocytic response have been reported and are evaluated by different tests. The following is a summary of the most important tests used to evaluate phagocytic function.
A. Neutrophil Count
This is the simplest and one of the most important tests to perform since phagocytic defects due to neutropenia are, by far, more common than the primary, congenital defects of phago-cytic function. As a rule, it is believed that a neutrophil count below 1000/ µL represents an increased risk of infection, and when neutrophil counts are lower than 200/ µL, the patient will invariably be infected.
B. Adherence
The increased adherence of activated phagocytic cells to endothelial surfaces is critical for the migration of these cells to infectious foci. Although specialized tests have been devel-oped to measure aggregation and adherence of neutrophils in response to stimuli such as C5adesarg (a nonchemotactic derivative of C5a), presently this property is evaluated indi-rectly by determining the expression of the different components of CD11/CD18 complex, which mediate adhesion by flow cytometry.
C. Chemotaxis and Migration
The migration of phagocytes in response to chemotactic stimuli can be studies in vitro us-ing the Boyden chamber. The basic principle of all versions of the Boyden chamber is to have two compartments separated by a membrane with pores too tight to allow PMN leuko-cytes to passively diffuse from one chamber to the other but large enough to allow the ac-tive movement of these cells from the upper chamber, where they are placed, to the lower chamber.
The movement of the cells is stimulated by adding to the lower chamber a chemo-tactic factor such as C5a, the bacterial tetrapeptide f-met-leu-phe, IL-8, leukotriene B4, or platelet-activating factor (PAF). The results are usually based on either counting the num-ber of cells that reached the bottom side of the membrane or on the indirect determination of the number of cells reaching the bottom chamber using51Cr-labeled PMN leukocytes (as illustrated in Fig. 13.3).
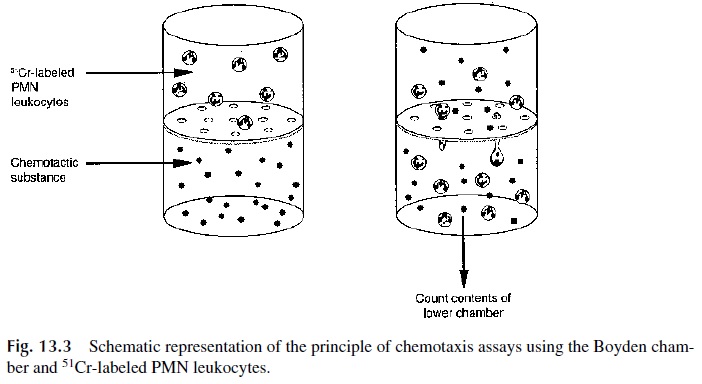
It must be noted that all versions of this technique are difficult to reproduce and stan-dardize and are not used in routine laboratory diagnosis.
D. Ingestion
Ingestion tests are relatively simple to perform and reproduce. They are usually based on incubating PMN with opsonized particles and, after an adequate incubation, determining either the number of ingested particles or a phagocytic index:
Phagocytic index = [No. of cells with ingested particles / Total no. of cells] × 100
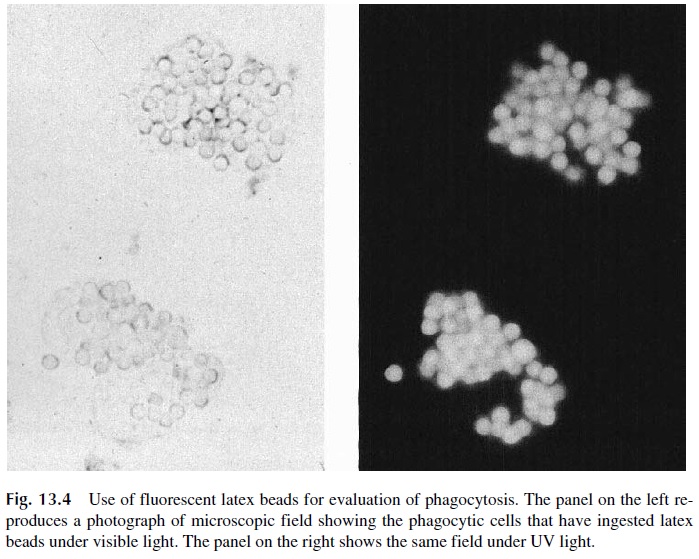
Several types of particles have been used, including latex, zymosan (fragments of fungal capsular polysaccharidic material), killed Candida albicans, and IgG-coated beads (Immunobeads). All these particles will activate complement by one of the pathways and become coated with C3, although opsonization with complement is not the major determi-nant of phagocytosis. The easiest particles to visualize once ingested are fluorescent latex beads; their use considerably simplifies the assay (Fig. 13.4), particularly if performed in a flow cytometer.
Ingestion tests are also not used routinely because other tests are available (e.g., the nitroblue tetrazolium reduction test; see below) that test both for ingestion and for the abil-ity to mount a respiratory burst.
E. Degranulation
When the contents of cytoplasmic granules are released into a phagosome, there is always some leakage of their contents into the extracellular fluid. The tests to study degranulation involve ingestion of particulate matter as mentioned above, but in this case the supernatants are analyzed for their contents of substances released by the PMN leukocyte granules such as myeloperoxidase, lysozyme, β-glucuronidase, and lactoferrin.
F. Measurement of the Oxidative Burst
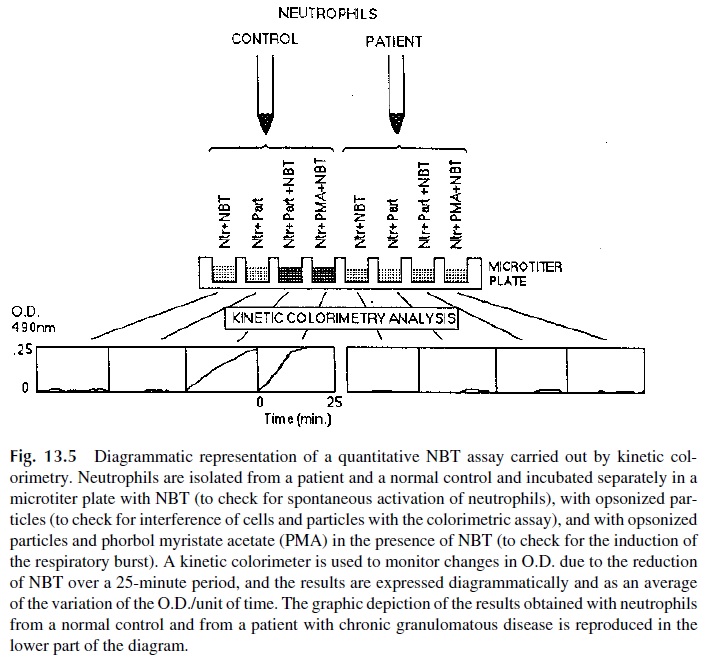
Most diagnostic laboratories that test for neutrophil function run a variant or another of a test to measure the oxidative burst. Several different assays are available, using dif-ferent parameters and methodologies. Tests based on the reduction of nitroblue tetra-zolium (NBT reduction tests) are the most commonly used for the evaluation of neu-trophil function.
The principle of the NBT assays is relatively simple. Oxidized NBT, colorless to pale yellow in solution, is transformed by reduction into blue formazan. The test usually in-volves incubation of purified neutrophils, NBT, and a stimulus known to activate the res-piratory burst. Two types of stimuli can be used:
1. Opsonized particles, which need to be ingested to stimulate the burst. In this way the test examines both the ability to ingest and the ability to produce a respiratory burst.
2. Diffusible activators, such as phorbol esters. Those compounds diffuse into the cell and activate protein kinase C, which in turn activates the NADPH– cytochrome B system and induce the respiratory burst directly, bypassing the ingestion step. A patient whose neutrophils respond to stimulation with phor-bol ester but not to stimulation with opsonized beads is likely to have an in-gestion defect. In contrast, neutrophils from a patient with a primary defect in the ability to generate the respiratory burst will not respond to any kind of stimulus.
The assessment of the ability of phagocytic cells to reduce NBT can be done micro-scopically or colorimetrically. The simplest NBT reduction assays rely on conventional mi-croscopy to count the number of PMN leukocytes with blue-stained cytoplasm after incu-bation with opsonized particles. However, the microscopic assay is difficult to standardize, and its interpretation can be affected by subjectivity.
The classical quantitative technique involves the extraction of intracellular NBT with pyrimidine and measure its absorbance at 515 nm (which corresponds to the absorbance peak of reduced NBT). This modality of the NBT test is extremely sensitive and accurate but is difficult to perform because the reagents used to extract the dye from the cells are highly toxic. An alternative are tests in which the PMN leukocytes are simultaneously ex-posed to opsonized particles and NBT and the change of color of the supernatant from pale yellow to gray or purple (as a result of the spillage of oxidizing products during phagocytosis) is measured. This assay was rendered practical and convenient by the introduction of kinetic colorimeters. Using this type of equipment the color change of NBT can be mea-sured without the need to extract the dye from the cells or to separate the cells from the supernatant (Fig. 13. 5).
The other techniques that are used in diganostic laboratories are based on the oxida-tion of 2’ ,7’ -dichlorofluorescein diacetate (nonfluorescent), which results in the formation of 2’ ,7’ -dichlorofluorescein (highly fluorescent). The numbers of fluorescent cells and flu-orescence intensity of activated and nonactivated PMN suspensions from patients and suit-able controls can be determined by flow cytometry. In patients with primary defects of the enzymes responsible for the respiratory burst, both the mean fluorescence intensity and the numbers of fluorescent cells after stimulation are considerably lower than those determined in normal, healthy volunteers.
G.Killing Assays
The main protective function of the neutrophil is the ingestion and killing of microorgan-isms. This ability can be tested using a variety of bacteria and fungi that are mixed with PMN leukocytes in the presence of normal human plasma (a source of opsonins), and after a given time the cells are harvested and lysed, and the number of intracytoplasmic viable bacteria is determined.
Killing assays are difficult and cumbersome and require close support from a mi-crobiology laboratory, and for this reason have been less used than the indirect killing as-says based on detection of the oxidative burst of the PMN leukocytes mentioned in the previous section. Alternative and simpler approaches to the evaluation of intracellular killing are based on the differential uptake of dyes (e.g., acridine orange) between live and dead bacteria.
Related Topics