Chapter: Essential Clinical Immunology: Immunological Aspects of Gastrointestinal and Liver Disease
Inflammatory Bowel Disease - Immune Mediated Diseases of the Gastrointestinal Tract
Inflammatory Bowel Disease
Crohn’s disease (CD) and ulcerative colitis (UC) are chronic relapsing and remitting inflammatory disorders of the gastroin-testinal tract, which together are referred to as IBD. Although clinical evidence sug-gests they are distinct entities, CD and UC are classified under the common name of IBD as they tend to occur in members of the same family and are thought to share genetic factors. While the exact pathoge-netic mechanisms remain to be uncovered, the current hypothesis is that IBD results from an inappropriate mucosal immune response to commensal flora in genetically predisposed hosts.
Much of what is known about CD and UC is the result of extensive study of mouse models of intestinal inflamma-tion that resemble IBD. The four general categories of such models are as follows:
(1) spontaneous colitis due to a naturally occurring genetic abnormality; (2) sponta-neous colitis in mice that have been geneti-cally manipulated – knockout and trans-genic mice; (3) colitis induced by exposure to a haptenating agent – an antigen that is immunogenic only upon being complexed with a carrier; and (4) adoptive trans-fer models that involve transfer of T-cell populations to a lymphopenic host such as mice with severe combined immunodefi-ciency (SCID).
Current research is focused on elu-cidating the complex interplay between genetic and environmental factors in the pathogenesis of IBD. Mouse models have allowed several broad conclusions to be made. The first, as demonstrated by the numerous murine models of colitis, is that different genetic defects can result in the same features of intestinal inflammation. The implication here is that there may be several mechanisms of IBD pathogenesis, which rely on a common pathway. Sec-ond, regardless of the genetic defect that is known to cause colitis, the host back-ground can largely determine whether inflammation occurs. One example is that IL-10 knockout mice bred onto different backgrounds express varying clinical phe-notypes. Careful study of these mice could provide insights about why some people develop IBD while others do not. Third, the normal luminal flora is an essential part of the development of colitis. Studies in several mouse models have demonstrated that colitis does not occur when mice are bred and maintained in a germ-free environment. However, once these mice are colonized by normal commensal flora, colitis rapidly ensues.
It is thought that IBD is the result of loss of tolerance to commensal flora and hence much interest is focused on uncovering the mechanisms that govern OT induction and the regulatory T-cell populations that are involved. By understanding OT, it may be possible to determine what defects occur in IBD, which would subsequently result in novel targets for immunotherapy.
CROHN’S DISEASE
In the United States, approximately 380,000 to 400,000 individuals are thought to be affected with CD. Even though it can occur at any age, a bimodal peak in the third and fifth decades of life has been observed. The incidence appears to be greater in whites than in blacks, and it has a predilection to affect the Ashkenazi-Jewish population.
There is a high concordance rate of 40–58 percent in monozygotic twins with CD. The use of microsatellite markers in genomewide screening of affected indi-viduals and family members has reaped the greatest benefits in terms of identify-ing susceptibility loci and genes that pre-dispose to the development of CD. There is a strong association with a single gene, NOD2, also referred to as CARD15 (caspase activation and recruitment domain) located on chromosome 16 and referred to as IBD1. NOD2 is an intracellular pattern recogni-tion receptor involved in the binding of muramyl dipeptide, the product of pep-tidoglycan derived from Gram-positive organisms. Of the three major mutations in NOD2 defined in CD, all result in the fail-ure of muramyl dipeptide binding and loss of NFκB activation.
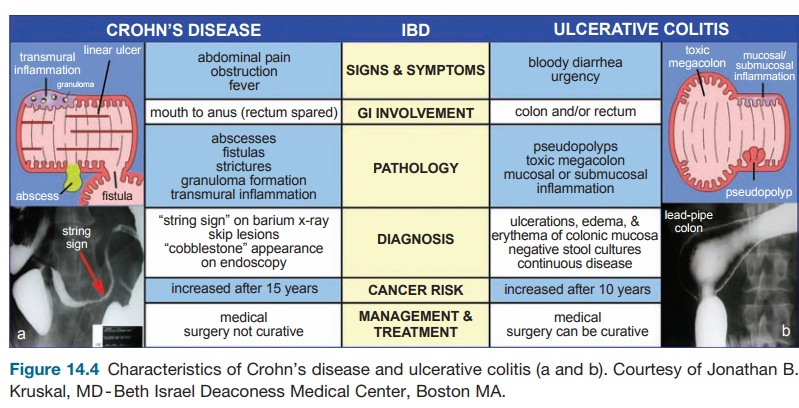
CD can involve any part of the GI tract, although it commonly affects the terminal ileum (30 percent), cecum and ileum (40 percent), perianal area (10 percent) and colon (20–30 percent). Endoscopy will often reveal diseased areas interspersed with normal areas of the bowel, called skip lesions. The nature of the inflamma-tory response leads to what is seen as a “cobblestone” appearance of the mucosa. This response is the result of linear ulcers intersecting with sections of normal tis-sue. Histologically, inflammation affects all the layers of the bowel wall (trans-mural). There is a dense infiltration of lymphocytes and macrophages and the presence of granulomas. Clinical features include abdominal pain, narrowing of the gut lumen (fibrosis) leading to strictures and bowel obstruction, abscess formation, fistulization to skin and internal organs, and if the colon is involved, diarrhea and bleeding (Figure 14.4).
Much evidence supports that CD is primarily a TH1-mediated immune response. However, recent studies impli-cated TH1 T-cells in the disease process. In mouse models, the introduction of the haptenating agent, trinitrobenzene sul-fonic (TNBS) acid, results in increased secretion of the cytokines IL-12, IFN-γ, and TNF-α. Histologic findings reveal a transmural inflammation in these mice that resembles that seen in CD. Increased secretion of IL-12 both in vivo and ex vivo by macrophages and dendritic cells isolated from patients with CD has also been observed. In CD patients, STAT-4 (signal transducer and activator of tran-scription–4) is increased in the bowel. IL-12 signals through STAT-4 to up-regulate IFN-γ. Antibodies specific for the p40 chain p of IL-12 have been shown to cause
Management of CD is generally accom-plished by medical rather than surgical means, although resection of affected bowel secondary to complications of dis-ease occurs in about 40 percent of patients in their lifetime. Surgery is rarely curative as the disease often recurs after resection. In fact, endoscopic recurrence occurs in about 90 percent of patients after one year, usually at the site of anastamosis. In part, decisions regarding medical management depend on the location, severity of disease, and extraintestinal complications. Therapy is targeted to two goals: (1) to treat acute flare-ups and (2) to maintain remission. Immunomodulators such as azathioprine, 6-mercaptopurine, and methotrexate are targeted toward maintaining remission. In recent years, the discovery of drugs tar-geting TNF-α, an inflammatory cytokine, has resulted in significant improvement in morbidity in individuals with CD who are refractory to other medications. A better understanding of the immunopathogenesis of CD can help establish new and effec-tive therapies. Emerging immunomodu-latory therapies that are currently being tested include antibodies to IL-12 and the mucosal addressin,α4β7.
ULCERATIVE COLITIS
In contrast to CD, histopathologic features and genetic factors are quite dif-ferent in UC. Monozygotic twin studies in patients with UC only demonstrated a 6–14 percent concordance rate. Although this rate is much lower than that for CD, common genetic loci suggest there is some influence of genetic factors on UC disease susceptibility as well.
Development of UC has been associ-ated with environmental factors. Appen-dectomy at a young age has been reported to have protective effects. The appendix is the site of abundant lymphoid tissue (PPs), and this may help generate protec-tive mucosal immune responses. Surpris-ingly, cigarette smoking is associated with decreased risk. One possible, although not proven, reason for this association is the effect of nicotine on mucin production and possibly pro-inflammatory cytokine secretion.
UC is thought to be mediated by a mixed inflammatory immune response that is generally characterized by the secretion of cytokines IL-5, IL-13, and IFN-γ. Thus, it is neither TH1 nor TH2. Mouse models of colitis in which the haptenating agent, oxa-zolone, is introduced as well as the TCRα −/− mouse demonstrate a similar cytokine profile.
UC only affects the colon and spreads proximally from the rectum in a con-tinuous fashion. In cases in which it only affects the rectum, it is calledulcerative proctitis. It affects the superficial layers of the bowel wall with infiltration of lympho-cytes, granulocytes, and mast cells. There are often ulcerations and crypt abscesses (collections of polymorphonuclear cells in the crypt). Clinically, patients often pres-ent with severe diarrhea, blood loss, and, with time and poor control, progressive loss of peristaltic function secondary to rigid “lead-pipe” colon. In severe cases, one can see the complications of this toxic megacolon and perforation. Of the IBDs, UC is strongly associated with an increased risk for colon cancer (occurs with long-standing – greater than ten years – disease) although this risk is present in CD as well (Figure 14.4).
Given that UC is generally confined to the colon, surgery is curative (total colec-tomy), in contrast to CD. Medical thera-pies include those that are used to treat CD – 5-ASA formulations, steroids, and immunomodulators. Surgery is reserved for those patients who are refractory to medications, develop intractable toxic megacolon, or show early or overt signs of colonic malignancy. The standard pro-cedure up until fifteen years ago was a total proctocolectomy and end ileostomy. However, more recently, a procedure that spares the rectal musculature and strips the rectal muscosa has been used, known as an ileoanal pull-through (restor-ative proctocolectomy with ileal pouch anal anastamosis). A total colectomy is performed, leaving the rectal muscula-ture layer that is essentially stripped of its mucosa, as the disease is superficial, and an internal pouch is created from the dis-tal ileum with outflow through the rectal muscular tube. A complication of this pro-cedure is inflammation of the pouch, or pouchitis. Interestingly, this complication occurs in UC patients in up to 40 percent of the cases but does not occur in individ-uals in which this procedure is performed for noninflammatory disease (e.g., famil-ial polyposis). These findings support an intrinsic defect in mucosal immunomo-regulation in UC.
Related Topics