Chapter: Basic & Clinical Pharmacology : Local Anesthetics
Historical Development of Local Anesthesia
Historical Development of Local Anesthesia
Although the numbing properties of cocaine were recognized for
centuries, one might consider September 15, 1884, to mark the “birth of local
anesthesia.” Based on work performed by Carl Koller, cocaine’s numbing effect
on the cornea was demonstrated before the Ophthalmological Congress in Heidelberg,
ushering in the era of surgical anesthesia. Unfortunately, with widespread use
came recognition of cocaine’s significant CNS and cardiac toxicity, which along
with its addiction potential, tempered enthusiasm for this application. As the
early investigator Mattison com-mented, “the risk of untoward results have
robbed this peerless drug of much favor in the minds of many surgeons, and so
deprived them of a most valued ally.” As cocaine was known to be a benzoic acid
ester, the search for alternative local anesthetics focused on this class of
compounds, resulting in the identification of benzocaine shortly before the
turn of the last century. However, benzocaine proved to have limited utility
due to its marked hydrophobicity, and was thus relegated to topical anesthesia,
a use for which it still finds limited application in current clinical
practice. The first useful injectable local anesthetic, procaine, was
introduced shortly thereafter by Einhorn, and its structure has served as the
template for the development of the most com-monly used modern local
anesthetics. The three basic structural elements of these compounds can be
appreciated by review of Table 26–1: an aromatic ring, conferring
lipophilicity, an ionizable tertiary amine, conferring hydrophilicity, and an
intermediate chain connecting these via an ester or amide linkage.
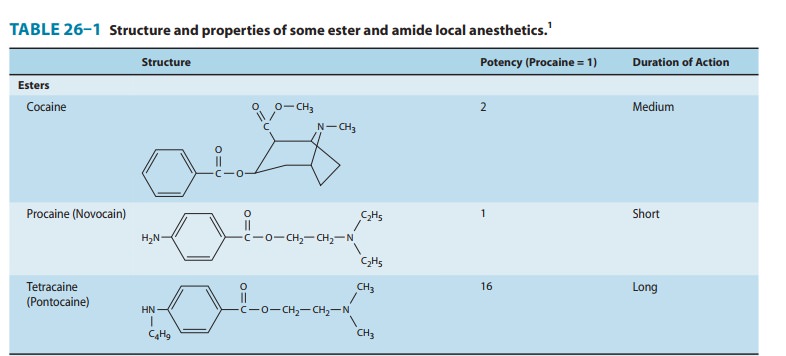
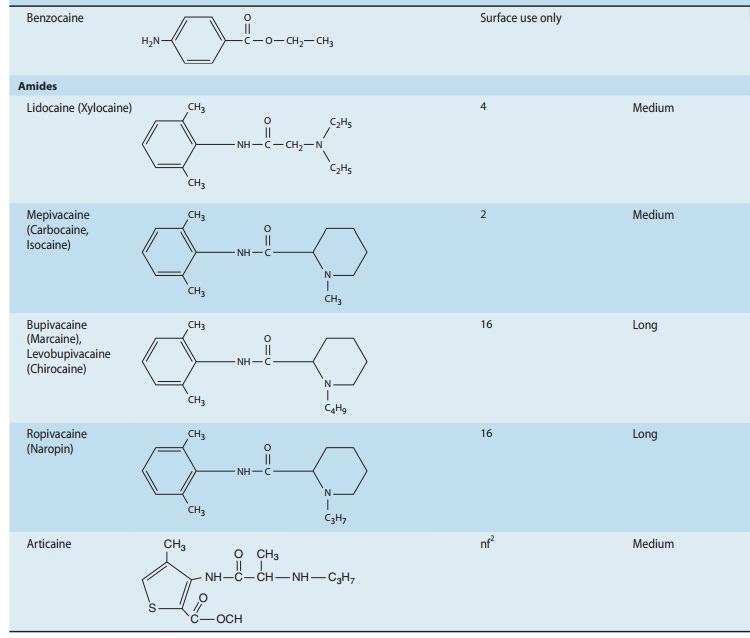
One of procaine’s limitations was its short duration of action,
a drawback overcome with the introduction of tetracaine in 1928. Unfortunately,
tetracaine demonstrated significant toxicity when employed for high-volume
peripheral blocks, ultimately reducing ts common usage to spinal anesthesia.
Both procaine and tetra-caine shared another drawback: their ester linkage
conferred insta-bility, and particularly in the case of procaine, the free
aromatic acid released during ester hydrolysis of the parent compound was
believed to be the source of relatively frequent allergic reactions.
Löfgren and Lundqvist circumvented the problem of instabil-ity
with the introduction of lidocaine in 1948. Lidocaine was the first in a series
of amino-amide local anesthetics that would come to dominate the second half of
the 20th century. Lidocaine had a more favorable duration of action than
procaine, and less sys-temic toxicity than tetracaine. To this day, it remains
one of the most versatile and widely used anesthetics. Nonetheless, some
applications required more prolonged block than that afforded by lidocaine, a
pharmacologic void that was filled with the intro-duction of bupivacaine, a
more lipophilic and more potent anes-thetic. Unfortunately, bupivacaine was
found to have greater propensity for significant effects on cardiac conduction
and func-tion, which at times proved lethal. Recognition of this potential for
cardiac toxicity led to changes in anesthetic practice, and sig-nificant
toxicity became sufficiently rare for it to remain a widely used anesthetic for
nearly every regional technique in modern clinical practice. Nonetheless, this
inherent cardiotoxicity would drive developmental work leading to the
introduction of two recent additions to the anesthetic armamentarium,
levobupiva-caine and ropivacaine. The former is the S(–) enantiomer
of bupi-vacaine, which has less affinity for cardiac sodium channels than its R (+) counterpart. Ropivacaine, another S(–) enantiomer, shares this reduced affinity for cardiac sodium
channels, while being slightly less potent than bupivacaine or levobupivacaine.
Related Topics