Chapter: Essentials of Psychiatry: Psychiatric Pathophysiology: Anxiety Disorders
Gamma-aminobutyric Acid (GABA)

Gamma-aminobutyric Acid (GABA)
GABA is the principal inhibitory transmitter in the
vertebrate central nervous system, with about one-third of all synapses using
GABA as a transmitter. Fifteen years after their introduction into clinical
practice, benzodiazepines were shown to augment the actions of GABA (Haefely
and Polc, 1986). A compelling body of evidence indicates that augmentation of
GABAergic transmission mediates the principal pharmacological actions of the
benzodiazepines. The efficacy of benzodiazepines in the treatment of anxiety
disorders (including GAD, PD and socialphobia) has been well documented by
double-blind, placebo-con-trolled studies (Ballenger, 1999; Blanco et al., 2002).
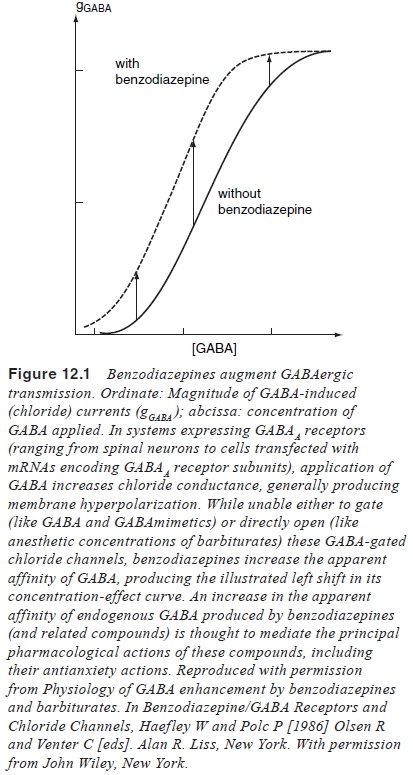
From a cellular perspective, benzodiazepines act as
allo-steric modulators, increasing the apparent affinity of GABA at GABAA
receptors (Figure 12.1). By binding to a specific subset of amino acids on the N-terminal of the GABAA
receptor alpha and gamma subunits (Wong et
al., 1992; Tretter et al., 1997),
ben-zodiazepines (and structurally unrelated compounds that bind to overlapping
sequences on these subunits) effect this increase in the apparent affinity of
GABA to its recognition site (Haefely and Polc, 1986).
Despite the evidence that benzodiazepines and other
strategies directed at augmenting GABAergic transmission are effective in the
treatment of anxiety disorders, this evidence does not directly implicate GABAA
receptors in their pathophysiol-ogy. Nonetheless, convergent pharmacological and
physiologi-cal evidence are consistent with the hypothesis that GABAergic
pathways are intimately involved in the behavioral, somatic and endocrine
manifestations of anxiety.
There are two principal lines of pharmacological
evidence implicating GABAergic pathways in anxiety. The first involves the
demonstration that so-called benzodiazepine receptor inverse agonists produce
a constellation of actions (behavioral, somatic and endocrine) reminiscent of anxiety in rodents and primates,
including man. Recall that the principal cellular action of ben-zodiazepines is
to increase the apparent affinity of GABA at GABAA receptors,
manifested as a left shift of the GABA con-centration effect curve (Figure
12.2).

The second line of pharmacological evidence linking
GABAA receptor dysfunction to anxiety disorders stems from reports
that flumazenil is panicogenic in PD patients but not in control subjects (Nutt
et al., 1990; Woods et al., 1991). It can be hypothesized
that flumazenil acts as a GABA negative (inverse agonist) compound in some
subsets of PD patients with an atypical
GABAA receptor composition in circumscribed areas of the central
nervous system (CNS). While there is no direct evidence to support this
hypothesis, positron emission tomography (PET) studies have demonstrated a
global reduction in the binding of 11C flumazenil throughout the brain in
patients with PD compared with controls.
Findings from multiple lines of research indicate that GABAA receptors may have evolutionary significance as a bio-warning system and perhaps as a means of compensating for perturbations in the environment. If stress-induced activation of GABAA receptors (manifested in these studies as increases in TBPS binding) is an attempt to compensate for environ-mental perturbations, then benzodiazepines (and low doses of barbiturates which increase the frequency of channel opening) (Study and Barker, 1981) may be viewed as pharmacological mimicry of the organism’s initial attempts to cope with the dis-ruption of its environment.
Related Topics