Chapter: Biochemical Pharmacology : Pharmacokinetics
Drug elimination: Metabolism
Drug elimination: Metabolism
While many drug moleculs can be eliminated
directly via the kidney, we have seen that others, predominantly hy-drophobic
ones, do not get efficiently secreted in the urine, be it because of plasma
protein binding or because of reup-take in the distal tubule. Even with some of
those drugs that are amenable to renal elimination, metabolism may occur and
give rise to changes in drug efficacy or to toxic side ef-fects. Drug metabolism
happens largely in the liver.
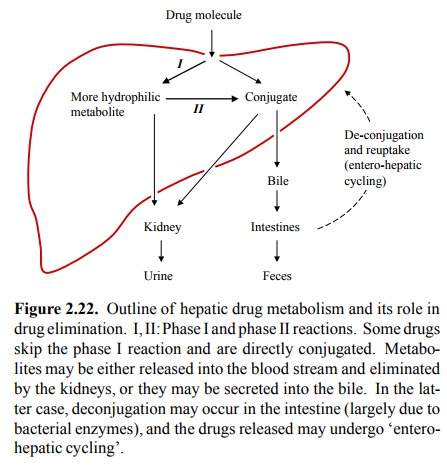
Drug metabolism is commonly –
and somewhat arbitrarily – subdivided into phase I and phase II reactions. In
Figure 2.22, phase I would correspond to the conversion of a drug molecule to a
more hydrophilic metabolite. The latter may then either be directly excreted in
the urine, or undergo conjugation with a larger polar moiety before excretion.
1. Example: Metabolism of phenobarbital and of morphine
We have seen above that
phenobarbital is not efficiently eliminated in the urine. It therefore is a
good candidate for elimination by hepatic metabolism. The molecule does not
have any good functional groups that could serve as points of attachment for
glucuronic acid or other polar moieties. Therefore, phenobarbital first has to
undergo a hydroxyla-tion reaction before conjugation may occur – an example of
a phase I reaction. Conjugation may then occur either with glucuronic acid, or
with sulfate (Figure 2.23a). Either mod-ification will inactivate the molecule
and render it suitable to renal excretion. The glucuronide may also be excreted
in the bile.
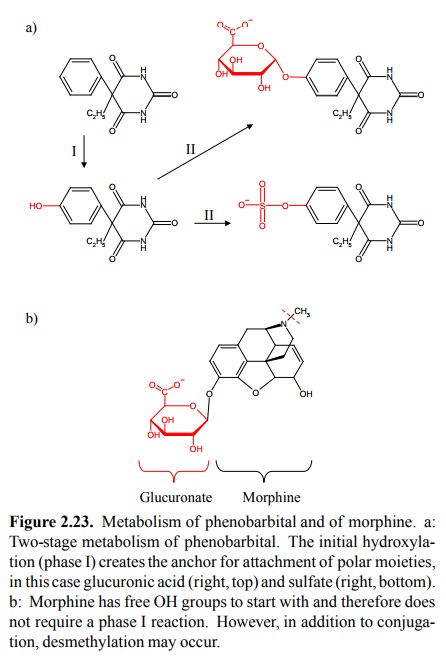
However, some drugs may not
undergo a phase I reaction at all but undergo conjugation directly. An example
is provid-ed my morphine2. Morphine has two free hydroxyl groups, to
either or both of which a UDP-glucuronosyltransferase in the liver ER will
attach a glucuronic acid moiety.
The conjugates formed with
glucuronic acid are called glu-curonides,
not glucuronates, because the bond created is a glycosidic bond but not an ester bond. The carboxylic acid group
remains free and contributes to the overall hy-drophilicity (Figure 2.23b).
2. Cytochrome P450 enzymes
Enzymes of the cytochrom P450
family are responsible for most phase I reactions. Cytochrome P450 enzymes are
extremely widespread in nature, and they occur in both prokaryotic and
eukaryotic cells. In eukaryotic cells, these enzymes mostly reside in the
membrane of the smooth en-doplasmic reticulum, but some variants are found in
the mi-tochondria.
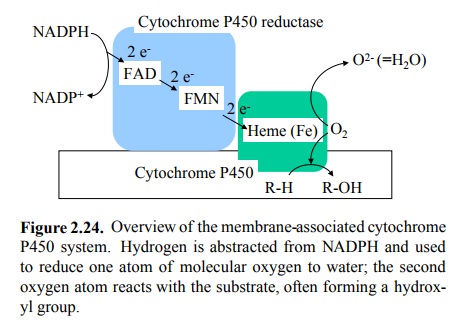
A cytochrome P450 enzyme
works in conjunction with a reductase, which supplies it with electrons from
NADPH (and uses FAD and FMN sequentially in the electron trans-fer process).
The two electrons are delivered to the heme cofactor in the active center of
the cytochrome, which in turn transfers them to one of the two oxygen atoms of
O2 to yield water (Figure 2.24). Presumably, the free energy of the
oxidation of NADPH is somehow utilized to facilitate the reaction of the other
oxygen with the organic substrate. This may result in the formation of a
phenolic hydroxyl group, as in the case of phenobarbital. However, the oxygen
may react with the substrate in various ways:
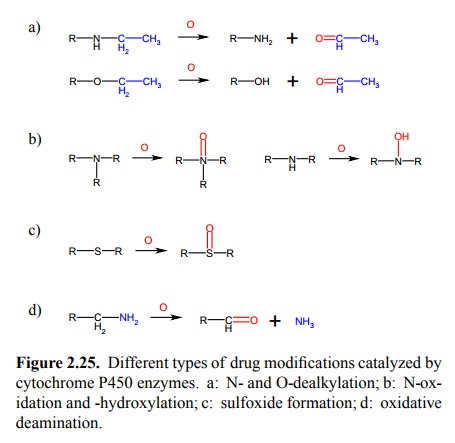
• N-dealkylation (Figure 2.25a; also see Fig.
2.23b)
• O-dealkylation (Figure 2.25a)
• N-oxidation and N-hydroxylation (Figure 2.25b)
• Sulfoxide formation (Figure 2.25c)
• Oxidative deamination (Figure 2.25d)
• Formation of epoxides from aromatic precursors.
This reaction may actually be quite harmful. Epoxides are highly reactive and
can do a lot of damage in the cell (more on this below).
The effects of cytochromes
P450 in drug metabolism are thus quite varied, and they involve numerous enzyme
species. However, it is noteworthy that one individual en-zyme– named CYP3A4 –
participates in the conversion of up to 60% of all drugs that do get
metabolized. While CYP3A4 is always present to some extent, the activity can be
substantially increased by a variety of drugs by a process called enzyme
induction. Basically, induction of drug-me-tabolizing enzymes works like the good,
old lac operon in Escherichia coli (Figure
2.26): The drug enters the cytosol and
associates with a protein receptor molecule named pregnane X receptor (PXR)
which is homologous to a num-ber of endogenous hormone receptors, many of which
bind steroid hormones.
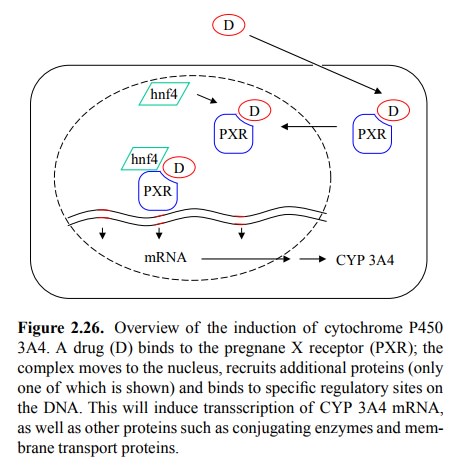
Upon drug binding, this
receptor translocates to the nucle-us, associates with some more proteins
(including hnf4) and binds to specific sites in the DNA to up-regulate sever-al
genes, including CYP3A4. Interestingly, it also induces membrane transporters
such as P-glycoprotein that are in-volved in excretion of metabolites from the
cell.
PXR has a remarkably broad
specificity, including both en-dogenous and exogenous ligands. Strong inducers
among clinically important drugs are phenytoin and phenobarbital (both used to
treat epilepsia), rifampicin (for tuberculosis), and ketoconazole (for fungal
infections). All these drugs are also substrates of CYP3A4, as are synthetic
steroid hormones used for contraception. This leads to a variety of clinically
important drug interaction phenomena: Oral contraception will cease to work
under treatment with ri-fampicin or phenytoin; dosages of phenytoin will have
to be increased during concomitant treatment with rifampicin, etc.
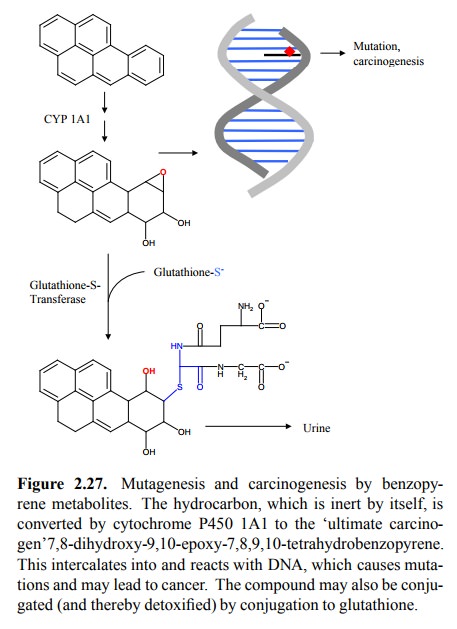
Another, homologous nuclear
receptor / transcriptional regulator called AHR (aromatic hydrocarbon receptor)
re-sponds to (surprise) aromatic hydrocarbons such asben-zopyrene, and it
induces the enzyme cytochrome P450 1A1. This enzyme will not only perform
hydroxylations but also introduce an epoxy group into the aromate. Polycyclic
aro-mates tend to `intercalate' between the base pairs of DNA, where the epoxy
group will react with some amino group, thus covalently fixing the damage in
the DNA (Figure 2.27). Although DNA repair mechanisms do exist, they are not
100% effective. Introduction of epoxy groups into ini-tially inert molecules
thus converts them into reactive ones that may potentially cause mutations and,
ultimately, can-cer. This reaction is not at all limited to liver tissue but is
ubiquitous; it very commonly occurs in the lungs. In fact, benzopyrene and
related compounds – formed during com-bustion of tobacco or the allegedly
indispensable wonder drug marijuana – are responsible for the induction of lung
cancer.
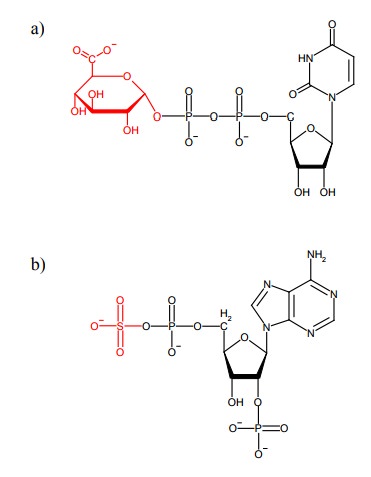
3. Overview of drug conjugation reactions
We have already seen a
variety of conjugation reactions in the foregoing examples. Important reactions
are
• Glucuronidation. These reactions are catalysed
by glu-curonosyltransferases and use the cosubstrate UDP-glu-curonic acid. The
glucuronate is most commonly trans-ferred to a hydroxyl group or to an amino
group.
• Acetylation. This is mediated by
acetyltransferases, uses acetyl-CoA and again mainly involves hydroxyl or amino
groups.
• Sulfation. Sulfotransferases use 3-phosphoadenosine-5-phosphosulfate
(PAPS) as a cosubstrate. It concerns mostly hydroxyl groups.
• Methylation. Methyltransferases use
S-Adenosylme-thionine as cosubstrate. Targets are hydroxyl, amino and
sulfhydryl groups.
• Glutathione conjugation. This is particularly
important with epoxides (Figure 2.27) but may also affects other functional
groups.
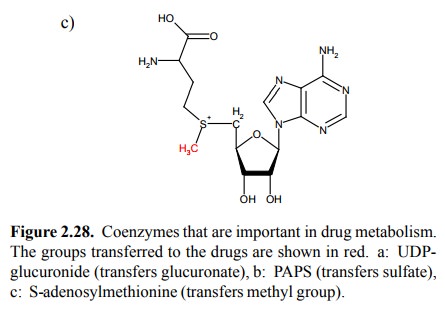
All the cosubstrates that
occur in drug conjugation (Figure 2.28) have other roles in metabolism; e.g.,
UDP-glucuron-ic acid and PAPS provide acidic groups for the synthesis of
mucopolysaccharides, whereas S-adenosylmethionine provides methyl groups for
the synthesis of phosphatidyl-choline from phosphatidylethanolamine.
Glucuronidation, sulfation
and glutathione conjugation will all increase the polarity of the drug substrate
and there-fore facilitate renal elimination. In contrast, methylation would not
seem to increase the polarity of the drug. Nor would it render the drug any
more amenable to further con-jugation; rather, the methyl group introduced
would tend to block reactive groups that could otherwise be used for the
attachment of glucuronic acid. What, then, is the `rationale' of methylation?
It may simply consist in the reduction or abolition of the drug's specific
activity by the change in its structure. The same would seem to apply to
acetylation, which utilizes the good, old acetyl-CoA as a cosubstrate, and is
shown in Fig. 2.30 for isoniazid (= isonicotinic acid hydrazide), a
tuberculostatic agent.
4. Glucuronidation
Phenolic or alcoholic
hydroxyl groups are the most com-mon functional groups to be conjugated with
glucuronic acid, as shown above for phenobarbital and morphine. Oth-er possible
sites of attachment include carboxylic acids, amines, hydroxylamines, and thiol
groups. This versatility is in keeping with the fact that glucuronidation is
the most common type of drug conjugation. With this modification, the dug
molecule acquires a negative charge and several hydroxyl groups, which will
render it considerably more polar and thus fit for excretion. Excretion may
happen ei-ther by way of the urine, or via the bile4. Hepatic
secretion (into the bile) works efficiently because all cells in the liver
tissue are not only connected to the blood vessels but also to capillary
tributaries of the bile duct. Glucuronides may be cleaved in the large
intestine by bacteria eager to metab-olize the glucuronic acid. One such
bacterium that possess-es glucuronidase is our good friend Escherichia coli. The released drug or metabolite may then be taken
up from the intestine again and then reach the liver, thus undergoing a
so-called enterohepatic cycle (Fig. 2.22, above). This ef-fect may result in
considerably delayed drug elimination. A practically important example is
digitoxin (discussed in the chapter on calcium), which is used in the treatment
of heart disease. The half-life of this drug extended to sever-al days because
of enterohepatic cycling. It is nevertheless often preferred over its analogue
digoxin (which is renal-ly eliminated) in those patients who have impaired
kidney function.
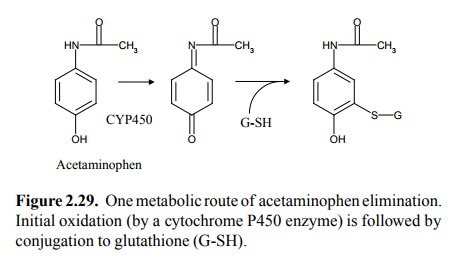
5. Glutathione conjugation
Glutathione is involved as a
reducing agent in a multiplic-ity of reactions in cell metabolism. Because of
its free sulfhydryl group, it is a very strong nucleophile, and be-cause of
that it is useful in the detoxification of the more difficult substrates such
as epoxides. Its depletion by drug conjugation may result in severe liver
damage. An exam-ple of such toxicity is acetaminophen, which at standard
dosages is a well-tolerated drug but is highly toxic to the liver at just 3 or
4 times that amount (Figure 2.29).
6. Acetylation
The `classical' model of drug metabolism by
acetylation is the tuberculostatic drug isoniacide (isonicotinic acid
hy-drazide). The metabolism of isoniazide has two interest-ing aspects:
Firstly, non-enzymatic hydrolysis of the acetyl metabolite releases
acetylhydrazine, which in turn is toxic. This, then, is an example of
detrimental drug metabolism (Figure 2.30a, b).
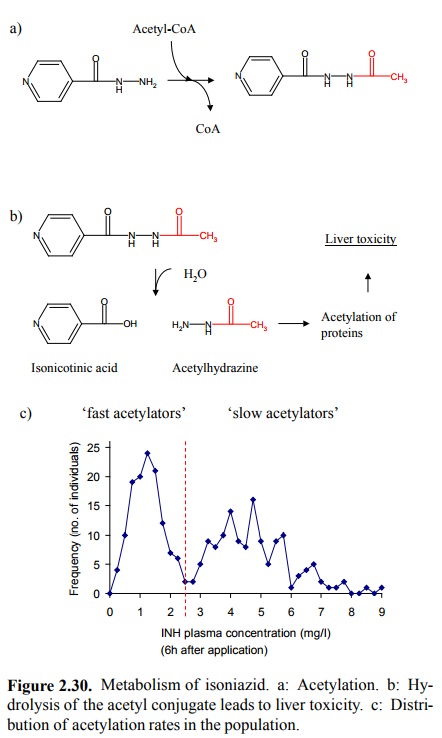
Secondly, the rate of the enzymatic acetylation
shows con-siderable inter-individual variation. This is illustrated in Fig.
2.30c. Shown are the plasma levels of unconjugated (i.e., not yet acetylated)
isoniazid in the plasma, 6 hours af-ter intake of a certain dosage of the drug.
The distribution is clearly
bimodal (which means, it has two separate peaks). People with a plasma level of
more than 2.5 mg/l are deemed `slow acetylators'. This is actually a genetic
trait that follows Mendelian inheritance, and it is obviously important for the
individual adjustment of isoni-azid dosage. It is the `classical' but by no
means single ex-ample of genetic variation in drug metabolism. The study of
phenomena of this type is called `pharmacogenetics', and there actually is a
scientific journal of that name.
7. Other reactions in drug metabolism
The conversions of prontosil
(azo reduction) and bacampi-cillin (ester hydrolysis), discussed in the
introduction, are examples of other important reactions in drug metabolism.
Related Topics