Chapter: Biochemical Pharmacology : Pharmacokinetics
Drug distribution - Pharmacokinetics
Drug distribution
Once the drug has entered the
systemic circulation, it needs to reach its target site. Target sites may be
located in vari-ous compartments:
1. Within the blood vessels. Example: blood
coagulation / clot dissolution. No problems of distribution here, drug
molecules of any size and shape can be used (when intravenously applied).
2. In the organ tissue, outside the blood vessels,
but extra-cellular or superficially exposed on the cell surface. Ex-ample: Most
receptors for hormones and transmitters.
3. In the organ tissue, intracellularly located.
Example: Many enzyme inhibitors.
1. Vascular permeability; the blood brain barrier
An important factor in the
distribution of drugs is the per-meability of the capillaries. Capillaries are
the microscop-ically small blood vessels across the very thin walls of which
metabolites and gases are exchanged between blood and tissues. Capillaries have
a cellular layer – the endothe-lium, supported by a basal membrane consisting
of proteins and proteoglycans (Figure microcirculation).
In the general circulation,
the endothelial cells have gaps between them (and sometimes fenestrations
across individ-ual cells, to the same effect). The permeability then is
de-termined by the sieving properties of the basal membrane, which permits
diffusion of salts, small molecules, and even some proteins, although most
plasma proteins are retained. This type of capillary does not present a barrier
to the dis-tribution of most drugs. However, in the brain and spinal chord (the
central nervous system, CNS), the endothelial cells are tightly connected by
structures called `tight junc-tions' and do not have fenestrations. In
addition, a second contiguous cellular layer is formed around the capillaries
by the glia cells. This adds up to four cell membranes layered in series – a
structure that is referred to as the blood
brain barrier. Therefore, even
small molecules cannot freely mi-grate into the brain tissue – or only so, if
they are extraordi-narily membrane-permeant.
Additional cell membrane
barriers (plasma membrane, and possibly organelle membranes) will have to be
overcome if the drug target is located intracellularly. It thus turns out that
cell membranes are of major importance as barriers toward drug distribution.

2. Drug hydrophobicity and permeation across membranes
Figure 2.6a shows the general
structure of a lipid bilayer (yawn). The obstacle to drug diffusion is the
hydrophobic core of the membrane. Substances that are lipophilic will traverse
the membrane more easily, because they readily partition into this hydrophobic
compartment (Figure 2.6b).
The lipid solubility of an
organic molecule is influenced in predictable ways by the functional groups it
contains. Charged and polar moieties will reduce lipid solubility, and
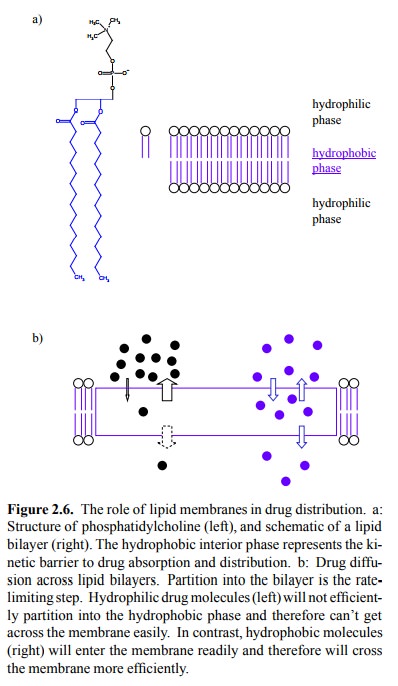
therefore render the drug molecules less membrane-perme-ant. Fig. 2.7a shows
some examples.
To increase lipid solubility, a drug may be
applied as a `pro-drug' that has some hydrophilic groups masked by more
hydrophobic ones. An example is bacampicillin, which is a derivative of the
antibiotic ampicillin (Figure 2.7b). Ester-ification of the carboxylic acid in
ampicillin facilitates up-take from the gut lumen. Esterases present in the
intestinal mucosal cells will cleave the ester and release ampicillin, which is
then passed on into the circulation.
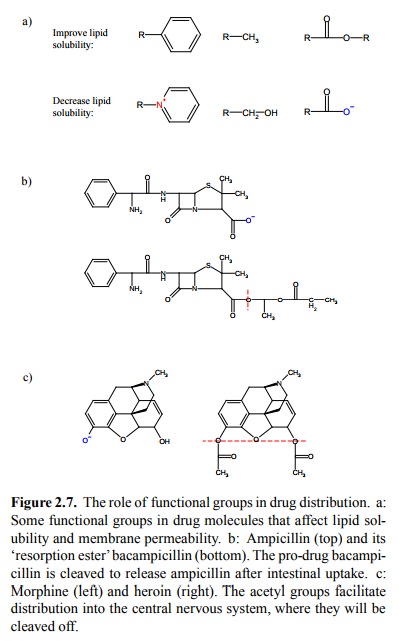
Masking hydrophilic groups also enhances the
uptake of drugs into the brain. A classical example is heroin, which is the
diacetylated derivative of morphine (Figure 2.7c). Ironically, heroin was
invented in an attempt to overcome the addictive effects of morphine. Methadone
was later invented to avoid those of heroin.
Another strategy to improve
the membrane permeant prop-erties of a drug is based on the effect of
`non-ionic diffu-sion'. An example is provided by the two `ganglion-block-ing'
agents hexamethonium and mecamylamine, which act as antagonists at certain
receptors of the transmitter acetyl choline (Figure 2.8a) and were formerly
used as antihyper-tensive agents. Acetylcholine is a quaternary amine; so is
hexamethonium. As a (dual) quaternary amine, hexametho-nium is not able to
traverse membranes and thus can only be applied intravenously. Mecamylamine,
however, is a ter-tiary amine and can adopt an uncharged (though
pharmaco-logically inactive) form that traverses membranes with ease. Having
reached its target site, it can change back into the charged form and exert its
effect. It can therefore be orally applied.
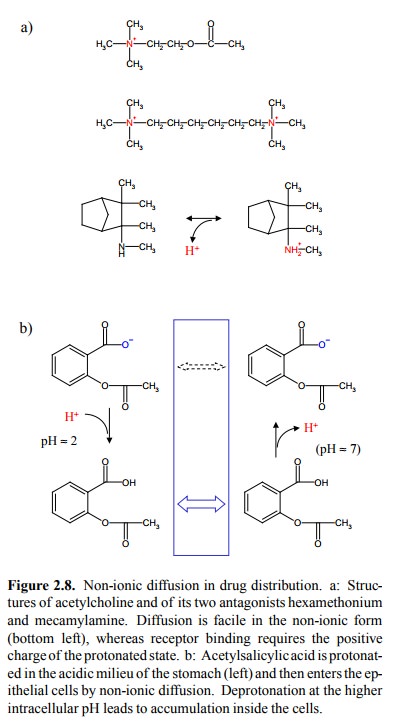
Non-ionic diffusion can also
produce unwanted effects, as in the case of aspirin (acetylsalicylic acid;
figure 2.8b). In the acidic milieu of the stomach, this molecule will be
protonated and thus uncharged, which promotes its diffu-sion into the cells of
the stomach mucous membrane. In-side the cell, the pH is very close to neutral,
which will lead to deprotonation of aspirin. Diffusion of the deprotonated
(charged) form out of the cell will be much slower than en-try, so that aspirin
will accumulate inside the cells to con centrations considerably higher than in
the stomach lumen. Aspirin, compared to other drugs that share its mechanism of
action (inhibition of cyclooxygenase; see later), has a stronger tendency to
trigger side effects such as gastritis and gastric or duodenal ulcera.
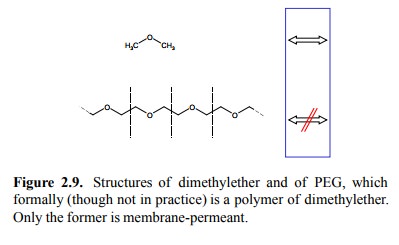
Molecular size is another
factor that is relevant to the ease of membrane permeation. This may be
illustrated by com-paring dimethylether (which crosses membranes readily) to
polyethyleneglycol, which may formally be considered a linear polymer of
dimethylether (Figure 2.9). PEG is quite efficiently excluded by membranes,
particularly in its high-er molecular weight varieties. It needs to be pointed
out, however, that this example is not entirely valid: PEG is not only larger
than dimethylether is but – for some subtle reason even our renowned polymer
chemist Jean Duhamel was not sure about either – it is also more polar.
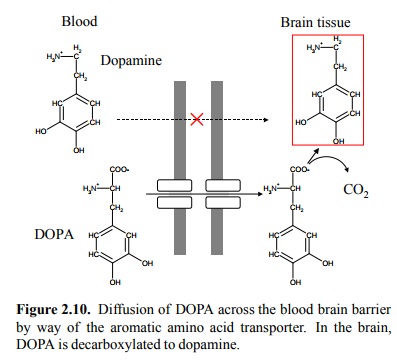
3. L-DOPA as an example of drug distribution facilitated by specific transport
Another strategy to overcome
membrane barriers is ex-emplified by DOPA (dihydroxyphenylalanine), the
precur-sor of dopamine (Figure 2.10). Dopamine is lacking in the brain in
Parkinson's disease. If dopamine itself is applied as a drug, it will not be
able to cross the blood brain bar-rier. Although its precursor DOPA is too
polar as well to cross the membrane by means of non-specific permeation, it can
take advantage of the limited specificity of the aro-matic amino acid
transporter. This transporter is found in the membranes that make up the blood
brain barrier, and its function consists in keeping the brain supplied with
pheny-lalanine, tyrosine, and tryptophan. Evidently, this strategy can be
applied only in exceptional cases.
4. The `volume of distribution'
After their uptake into the
systemic circulation, drugs are distributed between different compartments.
These compartments are usually summed up as follows (Figure 2.11a):
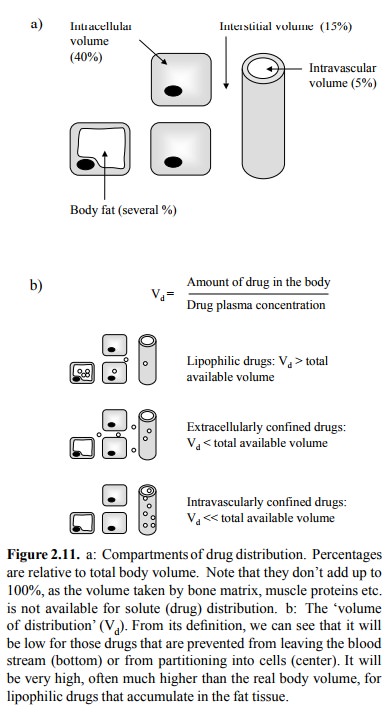
The `interstitial volume' is
the extracellular volume out-side of the blood vessels. Note that it is three
times larger than the intravascular volume! While its ionic composition closely
resembles that of blood plasma (with which it is in equilibrium for all small
solutes that are not protein-bound), it has a considerably lower protein
content.
Body fat is an important
reservoir for lipophilic drugs. This volume is more variable than the other
ones, so no general volume fraction can be given. However, values in the range
of 5-15% are not uncommon.
Few drugs are evenly
distributed among these compart-ments. Factors that will affect the equilibrium
distribution include:
• Membrane-impermeant drugs will be excluded from
the intracellular volume (Example: Lithium, which largely resembles sodium in
its distribution)
• Lipophilic drugs will be enriched in the fat
tissue (exam-ple: Thiopental – see later)
• Drugs with a high degree of protein binding
will be more enriched in the plasma (i.e., the intravascular vol-ume) than in
the interstitial fluid
An uneven distribution
between the intravascular and the (combined) extravascular spaces implies that
we cannot use the plasma concentration of a drug as an immediate mea-sure of
the total amount in the body. To correct for uneven distribution, a coefficient
named `volume of distribution' (Vd) has been invented (Figure
2.11a). This is not a real vol-ume but an experimentally determined number
(with the dimension of a volume, hence the fancy name).
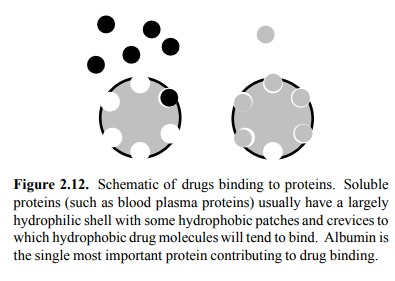
5. Protein binding
A factor that favours
retention of a drug in the intravascu-lar volume (at least in the short term)
is the binding of the drug to proteins (Figure 2.12), particularly albumin.
Pro-tein binding is usually more pronounced with hydrophobic drug molecules,
which are often bound to > 90% of their total concentration in the blood
plasma. Albumin is by far the most abundant single plasma protein. Moreover,
each albumin molecule affords multiple drug binding sites; these do not only
bind drugs but also fatty acids, which prevents toxic effects of the fatty acids
on cell membranes.
Protein binding is usually
rapidly reversible, so that the bound fraction is not `lost ' – it can yet
dissociate and bind to some drug target subsequently. However, one important
consequence of plasma protein binding is that it will pre- vent glomerular
filtration of the drug in the kidneys, which is an important step in drug
excretion (see below).
6. Kinetics of drug distribution
The above considerations on
drug partitioning mainly ap-ply to the equilibrium of drug distribution. However,
it is important to realize that it may take some time until a drug that is
applied rapidly (e.g., by injection or inhalation) ac-tually reaches
equilibrium. A practically important exam-ple of non-equilibrium distribution
is provided by the drug thiopental, which is a barbiturate used for
short-duration narcosis (Figure 2.13).
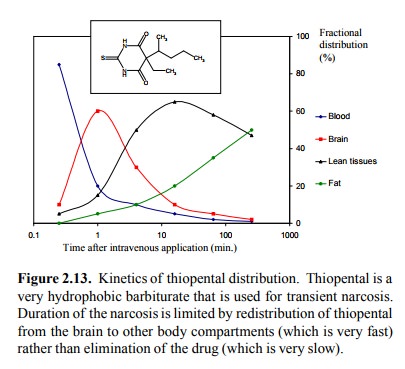
Thiopental is a very
lipophilic drug that readily crosses the blood brain barrier. Very shortly
after injection, the concentration in the brain peaks, and for a few minutes
the level is high enough to effect narcosis. This is due, among other things,
to the fact that the brain receives a very large fraction of the cardiac output
(~20%).
However, after a short time,
the drug leaves the brain again and accumulates in the lean tissues (such as
muscle), from where it finally redistributes to the body fat. This reflects
that the fat provides the most favourable (lipophilic) en-vironment; however,
since it is only weakly perfused, sub-stance exchange works more slowly than
with the other tis-sues. Note that, in this particular case, drug action is not
ter-minated by elimination of the drug (as is usually the case), but solely by
its redistribution from the site of action (the brain) to inert reservoirs
(muscle / fat). Ultimate elimina-tion is very slow – it takes days to complete
– and involves hepatic metabolism of the drug, followed by urinary ex-cretion.
Related Topics