Chapter: Modern Medical Toxicology: Asphyxiant Poisons: Toxic Gases
Carbon Monoxide - Systemic Asphyxiant Poison
SYSTEMIC ASPHYXIANTS
Carbon Monoxide
Synonyms
·
Carbonic oxide, Carbon oxide,
Exhaust gas, Flue gas.
Physical Appearance
·
Pure carbon monoxide is an
odourless, colourless, non-irritating gas, which is lighter than air.
Sources
· Incomplete combustion of almost any
form of fuel (wood, charcoal, gas, kerosene).
· Automobile exhaust.
· Fires.
· Paint remover (especially methylene
chloride).
· Tobacco smoke.
· Endogenous CO resulting from haeme
degradation can never reach toxic levels on its own. Normal CO level in plasma
is in the range of 1 to 5 % and may rise upto 7 to 8 % in smokers.
Usual Fatal Dose
This is usually expressed in terms of plasma concentration
of the gas (carboxyhaemoglobin or COHb). COHb level exceeding 50 to 60 % is
potentially lethal.
![]() A carbon
monoxide concentration of 5000 ppm in air is lethal to humans after five
minutes of exposure.
A carbon
monoxide concentration of 5000 ppm in air is lethal to humans after five
minutes of exposure.
Toxicokinetics
The lungs avidly absorb CO which combines with haemoglobin
(85%) and myoglobin (15%). Elimination occurs exclusively through the lungs.
Mode of Action
■■ Carbon monoxide has
an affinity for haemoglobin which is 230 to 270 times greater than that of
oxygen. Therefore, in spite of adequate partial pressure of oxygen (PO2)
in blood, there is reduced arterial oxygen content. Further, CO causes a leftward shift of the
oxyhaemoglobin dissocia-tion curve,* thus affecting the offloading of oxygen
from haemoglobin to the tissues. The net result of all this is the decreased
ability of oxygen to be carried by the blood and released to tissues.
■■ Apart
from the COHb-mediated hypoxia described, it is postulated that CO may also
interfere with cellular respiration by inactivating mitochondrial cytochrome
oxidase.
■■ CO
poisoning in experimental animals has been associ-ated with brain lipid peroxidation,
and thus a free radical peroxynitrate is produced which causes cellular
toxicity. In the brain this can cause further mitochondrial dysfunction,
capillary leakage, leukocyte sequestration and apoptosis. This change primarily
occurs during the recovery phase when lipid peroxidation occurs, which produces
an overall reversible demyelination in the brain. Common sites for CO-induced
brain injury are the basal ganglia, the cerebral white matter, hippocampus and
cerebellum.
■■ Cardiac
damage resulting in dysrhythmias is mainly because of reduced oxygen carrying
capacity of the blood due to COHb formation, and partially due to the binding
of CO with myoglobin.
■■ The
profound hypotension encountered in severe CO poisoning is due to 2 reasons:
activation of guanyl cyclase which relaxes smooth muscle, and displacement of
nitric oxide from platelets resulting in vasodilation.
■■ In a study on rats, the delayed effects of neuropathology following carbon monoxide poisoning were studied. The authors hypothesised that acute CO-mediated oxidative stress can cause alterations in myelin basic protein (a major myelin protein of the CNS), and that the immune response to these modified proteins can precipitate delayed neuro-logical dysfunction.
The results suggested that following CO
poisoning adduct formation between MBP and malony-laldehyde, a reactive product
of lipid peroxidation, causes an immunological cascade resulting in part in a
loss of antibody recognition of MBP. Thus, the neuropathology observed
following acute CO exposure may be linked to an adaptive immunological response
to chemically modified MBP. The authors suggested that these findings may have
clinical application in the treatment of delayed neurotox-icity with
anti-inflammatory agents.
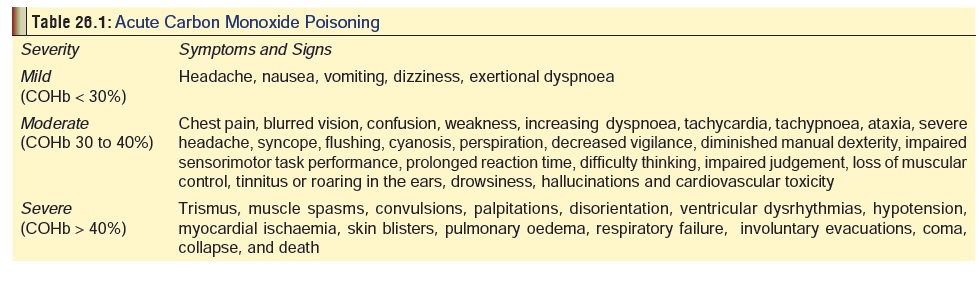
Clinical Features
Acute Exposure:
·
Mentioned in relation to severity of exposure in Table26.1. The earliest manifestations
are often non-specificand may be confused with other conditions. In fact
misdiagnosis is quite common unfortunately with CO exposure, especially in
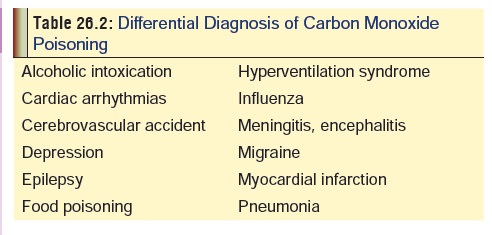
India where awareness about poisoning is generally low. Table 26.2 outlines the important conditions in the differential
diagnosis.
·
Two of the “classical” features of CO poisoning mentioned in
several textbooks on toxicolgy are actu-ally quite rarely encountered in
clinical practice:
o
Cherry red colour of blood and tissues (including skin) is
seen only in 2 to 3 % of cases.
o
Development of cutaneous bullae (blisters) is another
uncommon finding in clinical practice.
· It has been suggested that a more thorough examination of the eye (i.e. electrodiagnostic tests) would reveal that retinal haemorrhage may occur frequently, and that it can occur superficially or deeper in the nerve fibre layer (flame haemorrhage), and is often peripapillary. The venous changes that develop include engorgement and tortuosity, while oedema of the optic disc may be observed. All these changes reflect the hypoxic injury to the retina due to CO poisoning. Paracentral scotomata, homonymous hemianopia, tunnel vision, temporary blindness, and permanent blindness are known sequelae.
·
Although sensorineural hearing loss
is associated with acute CO poisoning, chronic low dose exposure to CO may
result in similar toxicity.
·
Myocardial ischaemia may be
precipitated or aggra-vated by CO; reported even with low CO levels in patients
with pre-existing coronary artery disease. Electrocardiographic changes of CO
poisoning include S-T segment depression or elevation, T wave abnormal-ities,
atrial fibrillation, and intraventricular conduction block.
·
Muscle necrosis, rhabdomyolysis, compartment syndrome and
elevated CPK have been reported following toxic exposures. Elevated CPK and
myoglo-binuria are characteristic. Delayed movement disor-ders have been
reported following CO poisoning. Haematuria, albuminuria, renal failure,
myoglobinuria, and acute tubular necrosis have developed with severe poisoning.
Lactic acidosis may occur.
·
Bullous lesions associated with carbon monoxide poisoning
generally appear within 24 hours of exposure and are usually located on the
palms and soles. They are not a common occurrence.
·
High susceptibility groups to CO poisoning include infants
(high respiratory and metabolic rates), preg-nant women, the elderly,
individuals with anaemia, haematologic disorders and patients with a history of
ischaemic heart disease or chronic obstructive lung disease. Children may be
more susceptible than adults to the neurological effects of CO, but no
statistical comparisons exist to support this claim.
·
A “post CO syndrome”, including headache, nausea, and
weakness may persist for 2 to 3 weeks following exposure to carbon monoxide.
Severe residual or delayed neurologic effects (“interval” form of CO poisoning)
may also occur after acute CO poisoning. Demyelination in the central nervous
system and other effects may occur 48 to 72 hours after exposure. The patient
should be observed carefully for CNS and other post-exposure hypoxic effects.
The most commonly involved regions of the brain include the globus pallidus and
the deep white matter. Signs and symptoms include mental deterioration,
irritability, aggressive behaviour, apathy, disorientation, hypokinesia,
akinetic mutism, distractibility, confusion, severe memory loss, delayed loss
of consciousness, coma, gait disturbances, faecal and urinary incontinence,
speech disturbances, tremor, bizarre behaviour, visual loss, movement
disorders, chorea, peripheral neuropathy, Tourette’s syndrome, and a
Parkinsonian syndrome. Physical findings include masked face, glabella sign,
grasp reflex, increased muscle tone, short stepped gait, retropulsion,
intention tremor, hyperreflexia, clonus, flaccid paresis, Babinski’s sign,
ataxia, and choreoathetosis.![]()
·
Another syndrome of delayed subtle neuropsychologic effects
has been described. Effects include headache, anorexia, nausea, apathy,
lethargy, forgetfulness, subtle personality changes and memory problems,
irritability and dizziness. These patients generally do not have gross
abnormalities on physical or neurologic exam. Neuropsychometric testing is
usually required to iden-tify abnormalities.
·
Recovery from the acute episode may be followed by permanent
neurological sequelae such as dementia, amnesia, psychosis, Parkinsonism,
paralysis, chorea, blindness, apraxia, agnosia, amnestic/confabulatory state,
depression, peripheral neuropathy, urinary/faecal inconti-nence, vegetative
state, and akinetic mutism. Personality changes may also occur, with increased
irritability, verbal aggression, violence, impulsivity and moodiness.
Chronic Exposure:
· The following features are seen
inchronically poisoned patients—
·
Headache, dizziness, confusion, intellectual deteriora-tion.
·
Weakness, nausea, vomiting, abdominal pain.
·
Paraesthesias
·
Visual disturbances: homonymous hemianopia, papil-loedema,
scotomata, retinal haemorrhages.
·
Hypertension, hyperthermia.
·
Cherry red skin.
·
Palpitations, aggravation of angina, intermittent
clau-dication.
·
Elevated RBC and WBC count.
·
Albuminuria, glycosuria.
·
Permanent neurological sequelae are common and include
amnesia, agnosia, apraxia, rigidity, personality changes, psychosis, blindness,
and hearing impairment.
·
CO exposure during pregnancy is teratogenic, depending upon
the stage of pregnancy. The foetus is more vulnerable to CO poisoning than the
mother. Exposure to the foetus can result in permanent brain damage, including
mental retardation, limb malfor-mation, hypotonia, areflexia, basal ganglia damage,
neuronal loss in the cerebral cortex, microcephalus, low infant birth weight,
telencephalic dysgenesis, seizures, and stillbirth.
Diagnosis
Summary—Determine COHb level when the
patient is firstseen and repeat every 2 to 4 hours until patient is asymptomatic,
or level is within the normal range. Monitor ECG, electrolytes, CPK,
urinalysis, arterial blood gases if symptomatic, or if the COHb level is
greater than 20%. Pulse oximetry may not provide a reliable estimate of
oxyhaemoglobin saturation.
· ![]() Estimation of carboxyhaemoglobin
level (COHb): Normal levels range from 0 to 5%, but in heavy smokers it may be
as high as 10%. The usual method of estima-tion is a co-oximeter, which
spectrophotometrically reads the percentage of total haemoglobin saturated with
CO. Either arterial blood or venous blood (in lithium heparin tube) can be
used. It must be borne in mind that COHb levels do not always correlate with
clinical manifestations or the final outcome.
Estimation of carboxyhaemoglobin
level (COHb): Normal levels range from 0 to 5%, but in heavy smokers it may be
as high as 10%. The usual method of estima-tion is a co-oximeter, which
spectrophotometrically reads the percentage of total haemoglobin saturated with
CO. Either arterial blood or venous blood (in lithium heparin tube) can be
used. It must be borne in mind that COHb levels do not always correlate with
clinical manifestations or the final outcome.
· Pulse oximetry: It is a non-invasive
method of measuring oxygen saturation and is relatively easy to perform,
painless, rapid, and accurate. A special sensor is placed on a patient’s
finger, toe, or nose. The sensor consists of a light-emitting diode that
projects two discrete wave-lengths of light corresponding to saturated and
unsatu-rated haemoglobin (660 and 940 nm) together with a photodetector.
Caution: In CO poisoning, pulse oximetry
gives higherreadings than the true HbO2 (oxyhaemoglobin) levels and
may fail to alert the physician to potentially lethal hypoxia. COHb absorbs
light almost identically to HbO2 at 660 nm. The oximeter responds to
COHb as if it were HbO2. Similarly the oximeter overestimates oxygen
satu-ration with increasing methaemoglobinaemia. A disparity between the oxygen
saturation calculated from PaO2 values and pulse oximetry readings
in fact should alert the physician to the presence of methaemoglobinaemia.
· Arterial blood gases: Partial
pressure of oxygen is usually normal, but the oxygen saturation expressed as a
percentage is decreased. A gap between the measured percentage HbO2
and the calculated percentage HbO2 indicates the necessity for
measuring COHb. PCO2 may be normal or slightly decreased. Metabolic
acidosis is invariably present.
· ECG: This may reveal myocardial
damage in the form of ST depression or elevation, T wave flattening or
inversion and dysrhythmias.
· Chest X-ray: This may reveal
ground-glass appearance, perihilar haze, peribronchial cuffing and
intra-alveolar oedema.
· CAT Scan: This may reveal
low-density globus pallidus lesions which are predictive of neurological
sequelae. Lucencies of the basal ganglia, particularly the globus pallidus is
characteristic of severe carbon monoxide poisoning. Low density lesions of
subcortical white matter, representing demyelination or necrosis, may also be
seen.
·
MRI: Cytotoxic oedema and demyelination, as well as damage
to white matter and basal ganglia are often detected accurately by MRI. In a
study of CO-poisoned patients, MRI scans performed 6 months after exposure
detected a 15 mm loss in the cross-sectional surface area of the corpus
callosum, compared with MRI images obtained on the day of CO exposure. The
effects appeared to be generalised atrophy, rather than sub-region specific
alterations. The authors suggested that long-term brain effects of CO poisoning
may be underestimated. T-2 weighted MRI may demonstrate abnormalities of the
basal ganglia, particularly the globus pallidus. Diffu-sion MRI has been used
as a more specific diagnostic aid following CO poisoning in some adults and
children following exposure.
·
Positron Emission Tomography (PET Scan): In a study of two
adults a few years after CO poisoning, PET scan imaging (findings indicated
significant metabolic decreases in the orbitofrontal and dorsolateral
prefrontal cortex as well as areas of the temporal lobe) was consis-tent with
the residual neurological deficits observed in each patient. The authors
suggested that PET imaging may be helpful in detecting the neuropathologic
sequelae associated with chronic nonlethal CO poisoning.
·
Ancillary Investigations:
·
Routine laboratory investigations often reveal elevated
serum creatine kinase and lactate dehydro-genase levels, as well as creatinine.
Hypokalaemia and hyperglycaemia are also usually present.
·
Neuropsychometric testing is indicated following
moderate-to-severe poisoning. Evaluated parameters included general
orientation, digit span, trailmaking, digit symbols, aphasia screening, and
block design. Equipment for doing this test include the WAIS set of nine blocks
for block design testing (8991-135).
·
Retinal haemorrhage is a common finding in CO poisoning. It
has been suggested that careful eye exam may provide useful diagnostic
information. Findings include superficial or deep retinal haemor-rhage, venous
changes (i.e. engorgement and tortu-osity) and oedema of the optic disc.
·
Bedside Tests:
o
Take 1 drop of blood and dilute with 10 to 15 ml of water.
Compare with normal blood diluted in the same manner. Blood containing carbon
monoxide is pink.
o
Add 0.1 ml of blood to 2 ml of ammonium hydroxide solution
(0.01 mol/L), and vortex-mix for 5 seconds. A pink tint in comparison with the
colour obtained from a normal blood specimen suggests the presence of COHb.
o
Dilute 1 ml of the patient’s blood with 10 ml of water in a
test tube and add to it 1 ml of a 5% solution of sodium hydroxide. If COHb is
present, the solution will turn straw yellow (< 20% COHb) or pink (> 20% COHb). In the case of normal
blood (HbO2) the solution turns brown in colour.
o
All the bedside tests are only screening tests and the
results must be confirmed by other methods mentioned earlier, especially
spectrophotometric estimation of COHb level.
Treatment
Admit
all patients with neurologic signs or symptoms, chest pain, abnormal EKG,
metabolic acidosis, and carboxyhaemo-globin level greater than 20%.
·
Immediate removal from the contaminated environment.
·
Oxygen (100%) through a tight-fitting mask or endo- tracheal
tube, until COHb falls to 15 to 20%. Onset of acute lung injury after toxic
exposure may be delayed up to 24 to 72 hours after exposure in some cases.
Maintain adequate ventilation and oxygenation with frequent monitoring of
arterial blood gases and/or pulse oximetry. If a high FIO2 is
required to maintain adequate oxygen-ation, mechanical ventilation and positive-end-expiratory
pressure (PEEP) may be required; ventilation with small tidal volumes (6 ml/kg)
is preferred if ARDS develops. Monitor cardiac and respiratory status.
·
Patients who only develop minor symptoms such as head- ache,
nausea and transient vomiting, who have normal mental status examinations and
neuropsychometric tests, and who are not pregnant may be treated with 100%
oxygen by non-rebreather mask and discharged when asymptomatic. Make sure
patients are not returning to a carbon monoxide contaminated environment.
·
Watch for the development of cerebral oedema with serial
neurologic exams, CAT scans, and fundoscopic examination. Hyperventilation (PCO
25 to 30 mmHg), head elevation (350), and mannitol 2(0.25 to 1 gm/Kg
of 20% solution over 30 minutes) are recommended as initial management of
raised intracranial pressure. The role of corticosteroids is controversial.
Refractory cere-bral oedema is due to cell death, and although mannitol, urea,
glycerol, or other methods to reduce life-threatening cerebral oedema may be
employed, they are unlikely to affect the outcome.
·
Metabolic acidosis must not be treated aggressively. Severe
acidosis should be treated. However, a slight acidosis may be beneficial by
shifting the oxygen-dissociation curve to the right, allowing more oxygen to be
released to the tissues. Therefore alkalaemia should be avoided. Sodium
bicarbonate is not recommended.
·
Administer supplemental glucose to prevent hypogly- caemia.
·
Convulsions can be controlled with IV diazepam or phenytoin
in the usual manner.
·
Physical activity should be restricted for at least 1 month
after the exposure to minimise the incidence of cerebral demyelination.
·
Antidote: Hyperbaric oxygen.
o
Several authorities consider administration of hyper- baric
oxygen (HBO) to be antidotal in its effects in carbon monoxide poisoning. It
involves inhalation of oxygen at a pressure greater than 1 atmosphere absolute
(ATA). 100% oxygen at ambient pressure reduces the half-life of COHb to 40
minutes, while at 2.5 atmospheres absolute it is reduced to just 20 minutes.
Hyperbaric oxygen should be instituted with 30 minutes of 100% oxygen at 3 ATA,
followed by 2 ATA for 60 minutes or until a COHb level less than 10% is
achieved.
o
HBO also increases the amount of dissolved oxygen by about
10 times which is an additional benefit. Further, animal studies indicate that
HBO prevents lipid peroxidation in the brain after loss of conscious-ness from
CO exposure, thereby minimising the incidence of neurologic damage. Studies
among human victims of CO poisoning indicate significantly reduced incidence of
neuropsychiatric symptoms in those treated with HBO as compared with those who
receive normobaric oxygen.
o
Normally a dramatic recovery of consciousness is seen during
hyperbaric treatment. Patients remaining unconscious may be given further
hyperbaric oxygen treatments.
o
It must be borne in mind however, that HBO therapy is
asociated with serious risks such as cerebral gas embo-lism, rupture of
tympanic membranes, visual deficits, reversible myopia, sweating, palpitations,
syncope, claustrophobia, and oxygen toxicity (convulsions and pulmonary
oedema). So the routine administration of HBO is not recommended in every case
of CO poisoning.
o
Severely ill patients should NOT be transferred to a
facility with a hyperbaric chamber until they have been stabilised: an airway
should be secured, venti-lation should be adequate, convulsions should be
controlled, and blood pressure and perfusion should be acceptable.
o
The decision to use hyperbaric oxygen during preg-nancy must
be based on several factors: The maternal need for HBO, the proven
foetotoxicity of CO, the theoretical foetotoxicity of HBO, and the absence of
demonstrated efficacy of HBO to prevent the foeto-toxicity of CO.
o
Table 26.3 lists the important indications for
HBOtherapy.

o Hyperbaric oxygen is also used in the treatment of poisoning due to cyanide, hydrogen sulfide, smoke, methylene chloride, and carbon tetrachloride.
Autopsy Features
· Cherry red (pink) colour of skin (Fig 26.2), especially noticeable in the
areas of postmortem lividity. In dark complexioned individuals, the colour can
be made out more easily in the inner aspects of lips, nail beds, tongue, and
palms and soles.

· Cutaneous bullae (skin blisters) are
sometimes seen in the regions of the calves, buttocks, wrists, and knees.
· Cherry pink colour of blood and
tissues. If blood is diluted with water in a test tube and held against light
or a white background, the pink colour will be more easily made out.
· Pulmonary oedema.
· The white matter of the brain is
said to be firmer than usual in CO poisoning, and the brain as a whole retains
its shape better after removal from the skull cavity.
· In a prospective study of
residential fire victims, soot deposits were monitored and were not found to be
predic-tive of CO poisoning. Although the absence of soot makes
carboxyhemoglobinaemia less likely, this study indi-cated that specificity was
low in determining actual CO poisoning.
· In delayed deaths, necrosis and
cavitation of basal ganglia, especially globus pallidus and putamen are
commonly described features. Petechiae and ring shaped haemorrhages may be seen
in the white matter. Heart may show focal areas of necrosis.
· It is mandatory to collect blood for
chemical analysis pref-erably from a peripheral vein. But unlike in other cases
of poisoning, if blood is difficult to obtain from a vein, heart blood or blood
from body cavities or even bone marrow can be used for analysis. Sodium
fluoride may be added as a preservative.
Forensic Issues
· Next to carbon dioxide, carbon monoxide is the most abun-dant atmospheric pollutant and is progressively increasing in concentration. Apart from its role as an environmental contaminant, CO is responsible for a significant number of deaths encountered in forensic practice. Once upon a time when domestic gas consisted of coal gas (which contained upto 7% CO), suicides accomplished with it at home were very common in Western countries. “Putting the head in the gas oven” was the most common form of self-destruction in countries such as the UK. Now that coal gas has been replaced by natural gas (which contains little or no CO), a major means of domestic suicide has been removed. But incomplete combustion of natural gas can release CO which can cause accidental poisoning in ill-ventilated areas.
·
Today the suicidal use of CO is utilised in a different way.
The victim utilises the exhaust fumes of a motor car either by merely sitting
in a closed garage with a window of the car open while the fumes build-up in
the enclosed area, or a device is fitted (e.g. a hose) to pipe the gas into the
inte-rior of the car with all windows rolled up. Such cases are however less
common in India and other Asian countries while they are quite frequently
reported in Western coun-tries. The use of catalytic converters in automobiles
has lessened the likelihood of death resulting from a suicide attempt via
inhalation of exhaust fumes.
·
Accidental CO poisoning can occur in several other situ-ations
apart from domestic exposure. Internal combus- tion engine exhaust fumes,
malfunctioning home heating systems, gas hot water heaters, gas clothes dryers,
charcoal and poorly vented wood/coal stoves, space heaters, gas and kerosene
lanterns, and fires in buildings are common sources of carbon monoxide
poisoning. Defective exhaust system of an automobile can allow gas to percolate
through the floor or engine bulkhead into the interior. Sometimes the driver
may become so affected that he loses control of the vehicle resulting in a
crash. The same applies to leakage of gas into the cockpit of a plane
(especially light aircraft) leading to the disablement of the pilot.
·
Tobacco smoke is an important source of carbon monoxide
contamination of environment. Mainstream cigarette smoke, that which is inhaled
into the smoker’s lungs, can contain as much as 5% carbon monoxide by volume.
Sidestream smoke, the source of environmental exposures, contains between 70
and 90% of the total CO per ciga-rette. In indoor areas where smoking is
permitted, carbon monoxide levels can exceed 11 ppm; this compares to less than
2 ppm in most non-smoking areas.
·
A common cause of accidental CO poisoning resulting in mass
deaths is a conflagration where-in a large building (hotel, theatre, block of
flats, etc.) goes up in flames. The majority of deaths in such cases are caused
by inhalation of smoke (containing CO) rather than by burns. A high- risk of CO
poisoning exists for fire fighters who often enter enclosed spaces in structural
fires. Use of respiratory protective gear can prevent lethal CO exposure, but
are not routinely used in all phases of fire fighting.
·
Homicidal poisoning with CO is rare, but cases have been
reported (and continue to be reported) from time to time.
·
Sudden infant death syndrome (SIDS) may be a misdiag-nosis
of carbon monoxide toxicity in some cases.
Related Topics