Chapter: Modern Pharmacology with Clinical Applications: Calcium Channel Blockers
Calcium Antagonism
CALCIUM
ANTAGONISM
The concept of calcium
antagonism as a specific mech-anism of drug action was pioneered by Albrecht
Fleckenstein and his colleagues, who observed that ve-rapamil and subsequently
other drugs of this class mimicked in reversible fashion the effects of Ca++
withdrawal on cardiac excitability. These drugs inhib-ited the Ca++ component
of the ionic currents carried in the cardiac action potential. Because of this
activity, these drugs are also referred to as slow channel block-ers, calcium channel antagonists, and calcium entry
blockers.
The actions of these drugs
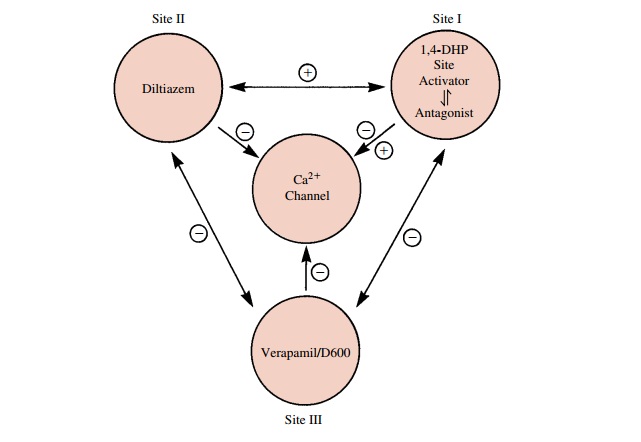
must be viewed from the perspective of cellular Ca++ regulation
(Fig. 19.1). Ca++ is fundamentally important as a messenger, linking
cel-lular excitation and cellular response. This role is made possible by the
high inwardly directed Ca++ concentra-tion and electrochemical
gradients, by the existence of specific high-affinity Ca++ binding
proteins (e.g., calmodulin) that serve as intracellular Ca++ receptors,
and by the existence of Ca++ -specific influx, efflux, and
sequestration processes. Calcium, in excess, serves as a mediator of cell
destruction and death during myo-cardial and neuronal ischemia, neuronal
degeneration, and cellular toxicity. The control of excess Ca++ mobi-lization
is thus an important contributor to cell and tis-sue protection.
The available Ca++ channel blockers exert their ef-fects
primarily at voltage-gated Ca++ channels of the plasma membrane. There are at least several
types of channels—L, T, N, P/Q and
R—distinguished by their electrophysiological and pharmacological
characteris-tics. The blockers act at the L-type channel at three dis-tinct
receptor sites (Fig. 19.2). These different receptor interactions underlie, in
part, the qualitative and quan-titative differences exhibited by the three
principal classes of channel blockers.
Cellular stimuli that involve Ca++ mobilization by processes other than that at the L-type voltage-gated channels will be either completely or relatively insensi-tive to the channel blockers. This differential sensitivity contributes to the variable sensitivity of vascular and nonvascular smooth muscle to the actions of these drugs, for example, the regional vascular selectivity and the general lack of activity of these agents in respiratory or gastrointestinal smooth muscle disorders.
The Selectivity of Action of Calcium Channel Blockers
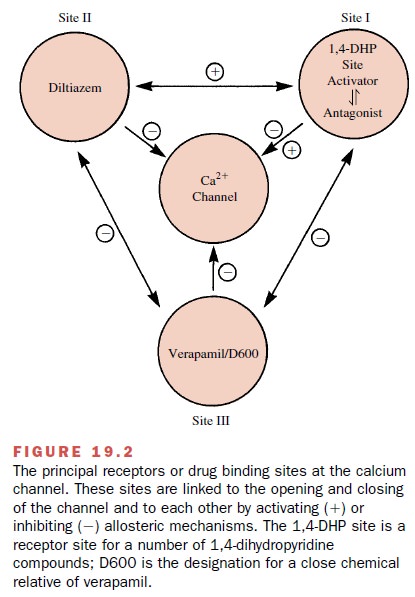
Although the available Ca++
channel blockers exert their effects through an interaction at one type of
chan-nel, they do so at different sites. Figure 19.2 shows that the channel
blockers act at three discrete receptor sites to mediate channel blockade
indirectly rather than by a direct or physical channel block. The existence of
the different receptor sites is one basis for the different pharmacological
profiles exhibited by these agents.
The activity of the Ca++
channel blockers increases with increasing frequency of stimulation or intensity
and duration of membrane depolarization. This
use-dependent activity is consistent with a preferred interac-tion of the
antagonists with the open or inactivated states of the Ca++ channel
rather than with the resting state. This
activity is not shared equally by all Ca++ blockers and so may
provide a further basis for the therapeutic dif-ferences between them. For
example, verapamil and diltiazem are approximately equipotent in cardiac and
vascular smooth muscle, whereas nifedipine and all other agents of the
1,4-dihydropyridine class are signifi-cantly more active in vascular smooth
muscle. Furthermore, different members of the 1,4-dihydropyri-dine class have
different degrees of vascular selectivity. These differences are broadly
consistent with the obser-vation that verapamil and diltiazem act
preferentially through the open channel state, and nifedipine and its analogues
act through the inactivated state.
The clinically available
calcium channel antagonists have also proved to be invaluable as molecular
probes with which to identify, isolate, and characterize calcium channels of
the voltage-gated family. In particular, the 1,4-dihydropyridines with their
high affinity, agonist– antagonist properties, and selectivity have become
de-fined as molecular markers for the L-type channel.
Synthetic drugs of comparable
selectivity and affin-ity to the 1,4-dihydropyridines do not yet exist for the
other channel types,T, N, P/Q, and R; these remain char-acterized by complex
polypeptide toxins of the aga- and conotoxin classes. Neuronal pharmacology,
including that of the central nervous system (CNS), is dominated by the N, P/Q,
and R channels. This underscores the normally weak effect of L-channel
antagonists on CNS function. Drugs that act at the N, P, and R channels with
comparable selectivity and affinity to the 1,4-dihydropyridines may be expected
to offer major po-tential for a variety of CNS disorders, including neu-ronal
damage and death from ischemic insults.
The Ca++ channel
blockers also differ in the extent of their additional pharmacological
properties. Verapamil and to a lesser extent diltiazem possess a number of
re-ceptor-blocking properties, together with NA+ and K+ channel–blocking
activities, that may contribute to their pharmacological profile. Nifedipine
and other 1,4-dihy-dropyridines are more selective for the voltage-gated Ca++
channel, but they may also affect other pharmaco-logical properties because
their nonpolar properties may lead to cellular accumulation. Together with
their channel-blocking properties, these properties may con- tribute to the
recently described antiatherogenic actions seen in experimental and clinical
states.
Related Topics