Chapter: Basic & Clinical Pharmacology : Cholinoceptor-Activating & Cholinesterase-Inhibiting Drugs
Basic Pharmacology of the Indirect Acting Cholinomimetics
BASIC PHARMACOLOGY
OF THE INDIRECT ACTING CHOLINOMIMETICS
The
actions of acetylcholine released from autonomic and somatic motor nerves are
terminated by enzymatic hydrolysis of the molecule. Hydrolysis is accomplished
by the action of acetylcholinest-erase, which is present in high concentrations
in cholinergic synapses. The indirect-acting cholinomimetics have their primary
effect at the active site of this enzyme, although some also have direct
actions at nicotinic receptors. The chief differences between members of the
group are chemical and pharmacokinetic—their pharmacodynamic properties are
almost identical.
Chemistry & Pharmacokinetics
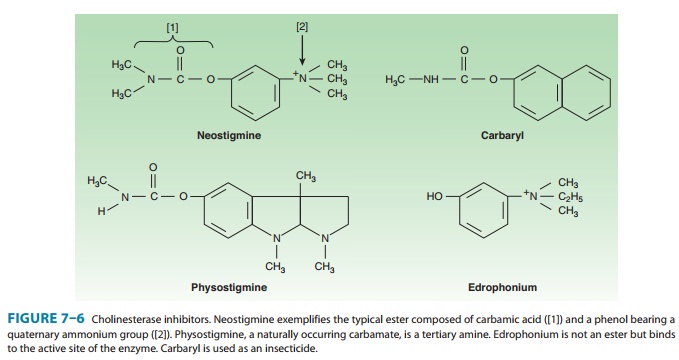
A. Structure
There are three chemical
groups of cholinesterase inhibitors:
(1) simple alcohols bearing a quaternary ammonium group, eg, edrophonium; (2)
carbamic acid esters of alcohols having quater-nary or tertiary ammonium groups
(carbamates, eg, neostigmine); and (3) organic derivatives of phosphoric acid
(organophosphates, eg, echothiophate). Examples of the first two groups are
shown in Figure 7–6. Edrophonium, neostigmine, and pyridostigmine are synthetic
quaternary ammonium agents used in medicine. Physostigmine (eserine) is a
naturally occurring tertiary amine of greater lipid solubility that is also
used in therapeutics. Carbaryl (carbaril) is typical of a large group of
carbamate insecticides designed for very high lipid solubility, so that
absorption into the insect and distribution to its central nervous system are
very rapid.
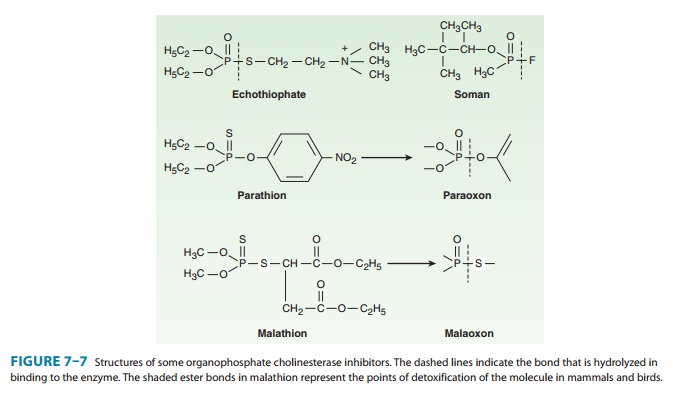
A
few of the estimated 50,000 organophosphates are shown in Figure 7–7. Many of
the organophosphates (echothiophate is an exception) are highly lipid-soluble
liquids. Echothiophate, a thio-choline derivative, is of clinical value because
it retains the very long duration of action of other organophosphates but is
more stable in aqueous solution. Soman is an extremely potent “nerve gas.”
Parathion and malathion are thiophosphate (sulfur-containing phosphate)
prodrugs that are inactive as such; they are converted to the phosphate
derivatives in animals and plants and are used as insecticides.
B. Absorption, Distribution, and Metabolism
Absorption
of the quaternary carbamates from the conjunctiva, skin, gut and lungs is
predictably poor, since their permanent charge ren-ders them relatively insoluble
in lipids. Thus, much larger doses are required for oral administration than
for parenteral injection. Distribution into the central nervous system is
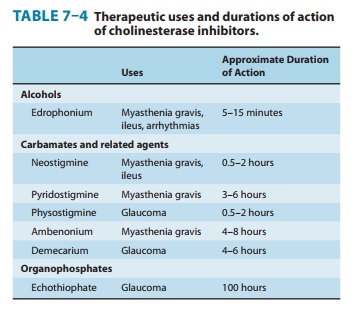
negligible. Physostigmine, in contrast, is well absorbed from all sites and can
be used topically in the eye (Table 7–4). It is distributed into the central
nervous system and is more toxic than the more polar quaternary carbamates. The
carbamates are relatively stable in aqueous solution but can be metabolized by
nonspecific esterases in the body as well as by cholinesterase. However, the
duration of their effect is deter-mined chiefly by the stability of the
inhibitor-enzyme complex (see Mechanism of Action, below), not by metabolism or
excretion.
The
organophosphate cholinesterase inhibitors (except for echothiophate) are well
absorbed from the skin, lung, gut, and conjunctiva—thereby making them
dangerous to humans and highly effective as insecticides. They are relatively
less stable than the carbamates when dissolved in water and thus have a limited
half-life in the environment (compared with another major class of
insecti-cides, the halogenated hydrocarbons, eg, DDT). Echothiophate is highly
polar and more stable than most other organophosphates. When prepared in
aqueous solution for ophthalmic use, it retains activity for weeks.
The
thiophosphate insecticides (parathion, malathion, and related compounds) are
quite lipid-soluble and are rapidly absorbed by all routes. They must be
activated in the body by conversion to the oxygen analogs (Figure 7–7), a
process that occurs rapidly in both insects and vertebrates. Malathion and a
few other organophosphate insecticides are also rapidly metabo-lized by other
pathways to inactive products in birds and mam-mals but not in insects; these
agents are therefore considered safe enough for sale to the general public.
Unfortunately, fish cannot detoxify malathion, and significant numbers of fish
have died from the heavy use of this agent on and near waterways. Parathion is
not detoxified effectively in vertebrates; thus, it is considerably more dangerous
than malathion to humans and livestock and is not available for general public
use in the USA.
All
the organophosphates except echothiophate are distributed to all parts of the
body, including the central nervous system. Therefore, central nervous system
toxicity is an important compo-nent of poisoning with these agents.
Pharmacodynamics
A. Mechanism of Action
Acetylcholinesterase
is the primary target of these drugs, but butyrylcholinesterase is also
inhibited. Acetylcholinesterase is an extremely active enzyme. In the initial
catalytic step, acetylcholine binds to the enzyme’s active site and is
hydrolyzed, yielding free choline and the acetylated enzyme. In the second
step, the cova-lent acetyl-enzyme bond is split, with the addition of water
(hydration). The entire process occurs in approximately 150 microseconds.
All the cholinesterase inhibitors increase the concentration of endogenous acetylcholine at cholinoceptors by inhibiting acetyl-cholinesterase. However, the molecular details of their interaction with the enzyme vary according to the three chemical subgroups mentioned above.
The
first group, of which edrophonium is the example, consists of quaternary
alcohols. These agents reversibly bind electrostatically and by hydrogen bonds
to the active site, thus preventing access of acetylcholine. The
enzyme-inhibitor complex does not involve a covalent bond and is
correspondingly short-lived (on the order of 2–10 minutes). The second group
consists of carbamate esters, eg, neostigmine and physostigmine. These agents
undergo a two-step hydrolysis sequence analogous to that described for
acetylcholine. However, the covalent bond of the carbamoylated enzyme is consid-erably more resistant to the second
(hydration) process, and this step is correspondingly prolonged (on the order
of 30 minutes to 6 hours). The third group consists of the organophosphates.
These agents also undergo initial binding and hydrolysis by the enzyme,
resulting in a phosphorylated active
site. The covalent phosphorus-enzyme bond is extremely stable and hydrolyzes in
water at a very slow rate (hundreds of hours). After the initial
binding-hydrolysis step, the phosphorylated enzyme complex may undergo a
process called aging. This process
apparently involves the breaking of one of the oxygen-phosphorus bonds of the
inhibitor and further strengthens the phosphorus-enzyme bond. The rate of aging
varies with the particular organophosphate compound. For example, aging occurs
within 10 minutes with the chemical warfare agent soman, but as much as 48
hours later with the drug VX. If given before aging has occurred, strong
nucleophiles like pralidoxime are able to break the phosphorus-enzyme bond and
can be used as “cholinesterase regenerator” drugs for organophosphate
insecticide poisoning . Once aging has occurred, the enzyme-inhibitor complex
is even more stable and is more difficult to break, even with oxime regenerator
compounds.
The
organophosphate inhibitors are sometimes referred to as “irreversible”
cholinesterase inhibitors, and edrophonium and the carbamates are considered
“reversible” inhibitors because of the marked differences in duration of
action. However, the molecular mechanisms of action of the three groups do not
support this simplistic description.
B. Organ System Effects
The
most prominent pharmacologic effects of cholinesterase inhibitors are on the cardiovascular
and gastrointestinal systems, the eye, and the skeletal muscle neuromuscular
junction (as described in the Case Study). Because the primary action is to
amplify the actions of endogenous acetylcholine, the effects are similar (but
not always identical) to the effects of the direct-acting cholinomimetic
agonists.
·
Central nervous system—In low concentrations,
the lipid-soluble cholinesterase inhibitors cause diffuse activation on the
electroencephalogram and a subjective alerting response. In higher
concentrations, they cause generalized convulsions, which may be followed by
coma and respiratory arrest.
·
Eye, respiratory tract,
gastrointestinal tract, urinary tract—The effects of the cholinesterase inhibitors
on these organsystems, all of which are well innervated by the
parasympatheticnervous system, are qualitatively quite similar to the effects
of the direct-acting cholinomimetics (Table 7–3).
·
Cardiovascular system—The cholinesterase
inhibitors canincrease activity in both sympathetic and parasympathetic ganglia
supplying the heart and at the acetylcholine receptors on neuro-effector cells
(cardiac and vascular smooth muscles) that receive cholinergic innervation.
In
the heart, the effects on the parasympathetic limb predomi-nate. Thus, cholinesterase
inhibitors such as edrophonium, phy-sostigmine, or neostigmine mimic the
effects of vagal nerve activation on the heart. Negative chronotropic,
dromotropic, and inotropic effects are produced, and cardiac output falls. The
fall in cardiac output is attributable to bradycardia, decreased atrial
con-tractility, and some reduction in ventricular contractility. The lat-ter
effect occurs as a result of prejunctional inhibition of norepinephrine release
as well as inhibition of postjunctional cel-lular sympathetic effects.
Cholinesterase
inhibitors have minimal effects by direct action on vascular smooth muscle
because most vascular beds lack cho-linergic innervation (coronary vasculature
is an exception). At moderate doses, cholinesterase inhibitors cause an
increase in sys-temic vascular resistance and blood pressure that is initiated
at sympathetic ganglia in the case of quaternary nitrogen compounds and also at
central sympathetic centers in the case of lipid-soluble agents. Atropine,
acting in the central and peripheral nervous systems, can prevent the increase
of blood pressure and the increased plasma norepinephrine.
The
net cardiovascular effects of
moderate doses of cholinest-erase inhibitors therefore consist of modest
bradycardia, a fall in cardiac output, and an increased vascular resistance
that results in a rise in blood pressure. (Thus, in patients with Alzheimer’s
disease who have hypertension, treatment with cholinesterase inhibitors
requires that blood pressure be monitored to adjust antihyperten-sive therapy.)
At high (toxic) doses of cholinesterase inhibitors, marked bradycardia occurs,
cardiac output decreases significantly, and hypotension supervenes.
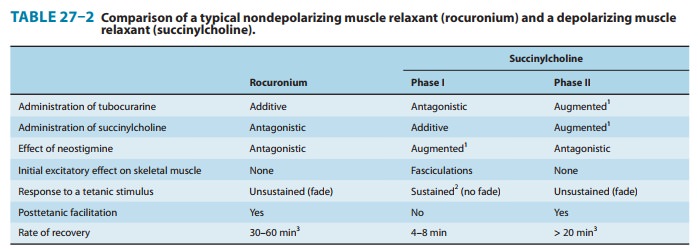
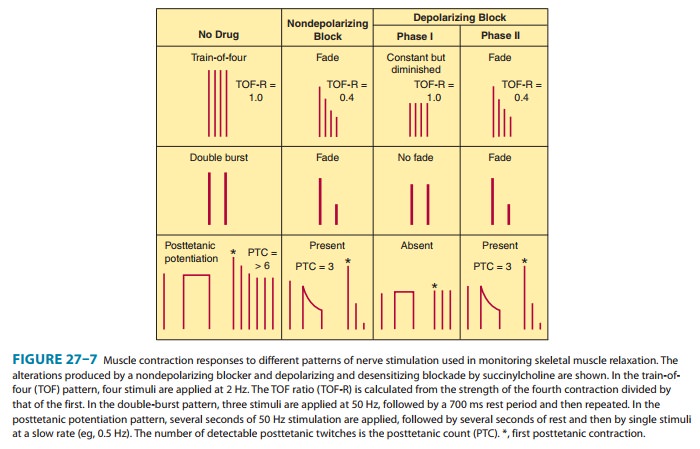
· Neuromuscular junction— The cholinesterase inhibitorshave important therapeutic and toxic effects at the skeletal muscle neuromuscular junction. Low (therapeutic) concentrations mod-erately prolong and intensify the actions of physiologically released acetylcholine. This increases the strength of contraction, especially in muscles weakened by curare-like neuromuscular blocking agents or by myasthenia gravis. At higher concentrations, the accumulation of acetylcholine may result in fibrillation of muscle fibers. Antidromic firing of the motor neuron may also occur, resulting in fasciculations that involve an entire motor unit. With marked inhibition of acetylcholinesterase, depolarizing neuromus-cular blockade occurs and that may be followed by a phase of nondepolarizing blockade as seen with succinylcholine (see Table 27–2 and Figure 27–7).
Some
quaternary carbamate cholinesterase inhibitors, eg, neo-stigmine, have an
additional direct nicotinic agonist
effect at the neuromuscular junction. This may contribute to the effectiveness
of these agents as therapy for myasthenia.
Related Topics