Chapter: Basic & Clinical Pharmacology : Cholinoceptor-Activating & Cholinesterase-Inhibiting Drugs
Basic Pharmacology of the Direct Acting Cholinoceptor Stimulants
BASIC
PHARMACOLOGY OF THE DIRECT ACTING CHOLINOCEPTOR STIMULANTS
The
direct-acting cholinomimetic drugs can be divided on the basis of chemical
structure into esters of choline (including acetyl-choline) and alkaloids (such
as muscarine and nicotine). Many of these drugs have effects on both receptors;
acetylcholine is typical. A few of them are highly selective for the muscarinic
or for the nicotinic receptor. However, none of the clinically useful drugs is
selective for receptor subtypes in either class.
Chemistry & Pharmacokinetics
A. Structure
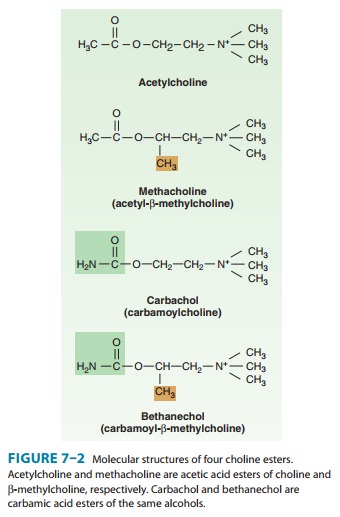
Four
important choline esters that have been studied extensively are shown in Figure
7–2. Their permanently charged quaternary ammonium group renders them
relatively insoluble in lipids. Many naturally occurring and synthetic
cholinomimetic drugs that are not choline esters have been identified; a few of
these are shown in Figure 7–3. The muscarinic receptor is strongly
stereo-selective: (S)-bethanechol is
almost 1000 times more potent than (R)-bethanechol.
B. Absorption, Distribution, and Metabolism
Choline
esters are poorly absorbed and poorly distributed into the central nervous
system because they are hydrophilic. Although all are hydrolyzed in the
gastrointestinal tract (and less active by the oral route), they differ
markedly in their susceptibility to hydrolysis by cholinesterase. Acetylcholine
is very rapidly hydrolyzed ; large amounts must be infused intravenously to
achieve concentrations sufficient to produce detectable effects. A large
intravenous bolus injection has a brief effect, typically 5–20 seconds, whereas
intramuscular and subcutaneous injections pro-duce only local effects.
Methacholine is more resistant to hydroly-sis, and the carbamic acid esters
carbachol and bethanechol are still more resistant to hydrolysis by
cholinesterase and have corre-spondingly longer durations of action. The β-methyl group (methacholine,
bethanechol) reduces the potency of these drugs at nicotinic receptors (Table
7–2).

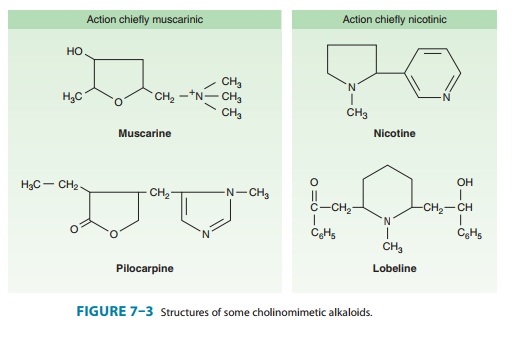
The
tertiary natural cholinomimetic alkaloids (pilocarpine, nicotine, lobeline;
Figure 7–3) are well absorbed from most sites of administration. Nicotine, a liquid,
is sufficiently lipid-soluble to be absorbed across the skin. Muscarine, a
quaternary amine, is less completely absorbed from the gastrointestinal tract
than the tertiary amines but is nevertheless toxic when ingested—eg, in certain
mushrooms—and it even enters the brain. Lobeline is a plant derivative similar
to nicotine. These amines are excreted chiefly by the kidneys. Acidification of
the urine accelerates clear-ance of the tertiary amines .
Pharmacodynamics
A. Mechanism of Action
Activation
of the parasympathetic nervous system modifies organ function by two major
mechanisms. First, acetylcholine released
Second, acetylcholine released from
parasympathetic nerves interacts with muscarinic receptors on nerve terminals
to inhibit the release of their neu-rotransmitter. By this mechanism,
acetylcholine release and circu-lating muscarinic agonists indirectly alter
organ function by modulating the effects of the parasympathetic and sympathetic
nervous systems and perhaps nonadrenergic, noncholinergic (NANC) systems.
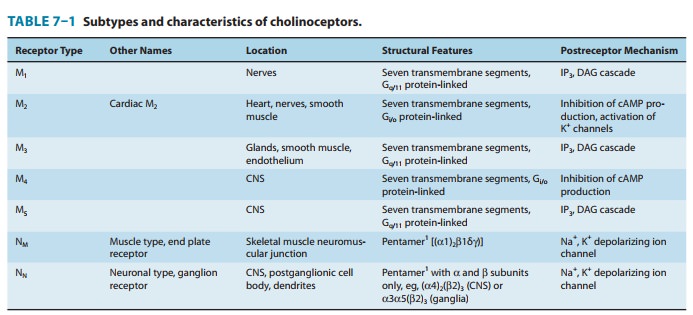
As
indicated, muscarinic receptor subtypes have been characterized by binding
studies and cloned. Several cellular events occur when muscarinic receptors are
activated, one or more of which might serve as second messengers for muscarinic
activa-tion. All muscarinic receptors appear to be of the G protein-coupled
type (Table 7–1). Muscarinic agonist binding activates the inositol
trisphosphate (IP3), diacylglycerol (DAG) cascade. Some evidence
implicates DAG in the opening of smooth muscle calcium channels; IP3
releases calcium from endoplasmic and sarcoplasmic reticulum. Muscarinic
agonists also increase cellular cGMP concentrations. Activation of muscarinic
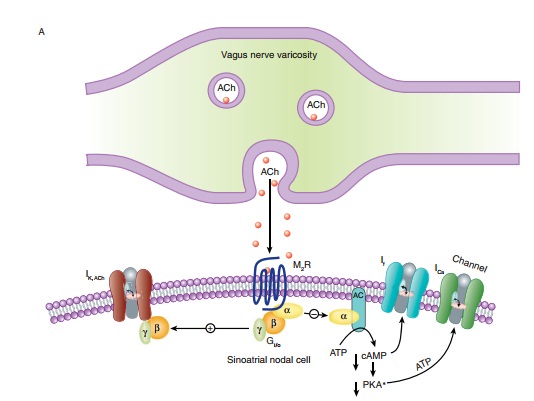
receptors also increases potassium flux across cardiac cell membranes (Figure
7–4A) and decreases it in ganglion and smooth muscle cells. This effect is
mediated by the binding of an activated G protein βγ subunit directly to the channel. Finally,
muscarinic receptor activation in some tissues (eg, heart, intestine) inhibits
adenylyl cyclase activity. Moreover, muscarinic agonists attenuate the
activation of adenylyl cyclase and modulate the increase in cAMP levels induced
by hormones such as catecholamines. These muscarinic effects on cAMP generation
reduce the physiologic response of the organ to stimulatory hormones.
The
mechanism of nicotinic receptor activation has been stud-ied in great detail,
taking advantage of three factors: (1) the recep-tor is present in extremely
high concentration in the membranes of the electric organs of electric fish;
(2) α-bungarotoxin,
a com-ponent of certain snake venoms, binds tightly to the receptors and is
readily labeled as a marker for isolation procedures; and receptor activation
results in easily measured electrical and ionic changes in the cells involved.
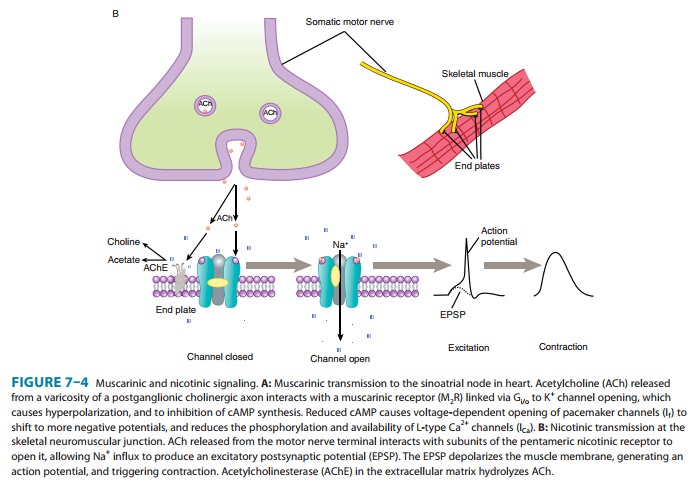
The nicotinic receptor in muscle tissues is a pentamer of four types of
glycoprotein sub-units (one monomer occurs twice) with a total molecular weight
of about 250,000 (Figure 7–4B). The neuronal nicotinic recep-tor consists of α and β subunits only (Table
7–1). Each subunit has four transmembrane segments. The nicotinic receptor has
two agonist binding sites at the interfaces formed by the two α subunits and two
adjacent subunits (β,
γ,
ε).
Agonist binding to the receptor sites causes a conformational change in the
protein (channel opening) that allows sodium and potassium ions to dif-fuse
rapidly down their concentration gradients (calcium ions may also carry charge
through the nicotinic receptor ion channel). Binding of an agonist molecule by
one of the two receptor sites only modestly increases the probability of
channel opening; simultaneous binding of agonist by both of the receptor sites
Nicotinic receptor activation causes
depolarization of the nerve cell or neuromuscular end plate membrane. In
skeletal muscle, the depolarization initiates an action potential that
propagates across the muscle membrane and causes contraction (Figure 7–4B).
Prolonged
agonist occupancy of the nicotinic receptor abolishes the effector response;
that is, the postganglionic neuron stops firing (ganglionic effect), and the
skeletal muscle cell relaxes (neuromus-cular end plate effect). Furthermore,
the continued presence of the nicotinic agonist prevents electrical recovery of
the postjunctional membrane. Thus, a state of “depolarizing blockade” occurs
initially during persistent agonist occupancy of the receptor. Continued
agonist occupancy is associated with return of membrane voltage to the resting
level. The receptor becomes desensitized to agonist, and this state is
refractory to reversal by other agonists.
B. Organ System Effects
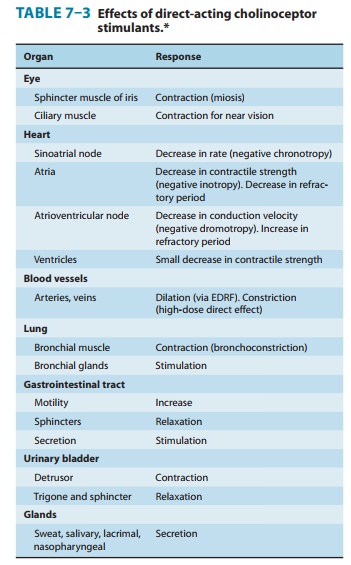
Most
of the direct organ system effects of muscarinic cholinoceptor stimulants are
readily predicted from knowledge of the effects of parasympathetic nerve
stimulation (see Table 6–3) and the distri-bution of muscarinic receptors.
Effects of a typical agent such as acetylcholine are listed in Table 7–3. The
effects of nicotinic ago-nists are similarly predictable from knowledge of the
physiology of the autonomic ganglia and skeletal muscle motor end plate.
1.
Eye—Muscarinic agonists
instilled into the conjunctival saccause contraction of the smooth muscle of
the iris sphincter (result-ing in miosis) and of the ciliary muscle (resulting
in accommoda-tion). As a result, the iris is pulled away from the angle of the
anterior chamber, and the trabecular meshwork at the base of the ciliary muscle
is opened. Both effects facilitate aqueous humor outflow into the canal of
Schlemm, which drains the anterior chamber.
2.
Cardiovascular system—The primary
cardiovasculareffects of muscarinic agonists are reduction in peripheral
vascular resistance and changes in heart rate. The direct effects listed in
Table 7–3 are modified by important homeostatic reflexes, as described and
depicted in Figure 6–7. Intravenous infusions of minimally effective doses of
acetylcholine in humans (eg, 20–50 mcg/min) cause vasodilation, resulting in a
reduction in blood pressure, often accompanied by a reflex increase in heart
rate. Larger doses of acetylcholine produce bradycardia and decrease
atrioventricular node conduction velocity in addition to hypotension.
The
direct cardiac actions of muscarinic stimulants include the following: (1) an
increase in a potassium current (IK(ACh)) in the cells of the
sinoatrial and atrioventricular nodes, in Purkinje cells,
and
also in atrial and ventricular muscle cells; (2) a decrease in the slow inward
calcium current (ICa) in heart cells; and (3) a reduc-tion in the
hyperpolarization-activated current (If ) that underlies diastolic
depolarization (Figure 7–4A). All these actions are medi-ated by M2
receptors and contribute to slowing the pacemaker rate. Effects (1) and (2)
cause hyperpolarization, reduce action potential duration, and decrease the
contractility of atrial and ventricular cells. Predictably, knockout of M2
receptors eliminates the bradycardic effect of vagal stimulation and the
negative chro-notropic effect of carbachol on sinoatrial rate.
The
direct slowing of sinoatrial rate and atrioventricular con-duction that is
produced by muscarinic agonists is often opposed by reflex sympathetic
discharge, elicited by the decrease in blood pressure (see Figure 6–7). The
resultant sympathetic-parasympathetic interaction is complex because muscarinic
modulation of sympa-thetic influences occurs by inhibition of norepinephrine
release and by postjunctional cellular effects. Muscarinic receptors that are
present on postganglionic parasympathetic nerve terminals allow neurally
released acetylcholine to inhibit its own secretion. The neuronal muscarinic
receptors need not be the same subtype as found on effector cells. Therefore,
the net effect on heart rate depends on local concentrations of the agonist in
the heart and in the vessels and on the level of reflex responsiveness
Parasympathetic
innervation of the ventricles is much less extensive than that of the atria;
activation of ventricular muscar-inic receptors causes much less physiologic
effect than that seen in atria. However, the effects of muscarinic agonists on
ventricular function are clearly evident during sympathetic nerve stimulation
because of muscarinic modulation of sympathetic effects (“accen-tuated
antagonism”).
In
the intact organism, intravascular injection of muscarinic agonists produces
marked vasodilation. However, earlier studies of isolated blood vessels often
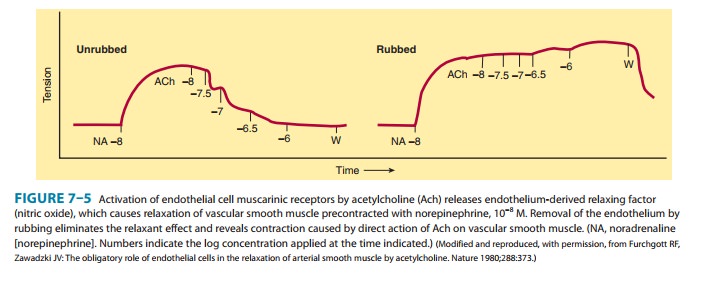
showed a contractile response to these agents. It is now known that
acetylcholine-induced vasodi-lation arises from activation of M3
receptors and requires the presence of intact endothelium (Figure 7–5).
Muscarinic ago-nists release endothelium-derived relaxing factor (EDRF),
iden-tified as nitric oxide (NO), from the endothelial cells. The NO diffuses
to adjacent vascular smooth muscle, where it activates guanylyl cyclase and
increases cGMP, resulting in relaxation (see Figure 12–2). Isolated vessels prepared
with the endothelium preserved consistently reproduce the vasodilation seen in
the intact organism. The relaxing effect of acetylcholine was maximal at 3 × 10−7 M
(Figure 7–5). This effect was eliminated in the absence of endothelium, and
acetylcholine, at concentrations greater than 10−7 M, then caused
contraction. This results from a direct effect of acetylcholine on vascular
smooth muscle in which activation of M 3 receptors stimulates IP3
production and releases intracellular calcium.
Parasympathetic
nerves can regulate arteriolar tone in vascular beds in thoracic and abdominal
visceral organs. Acetylcholine released from postganglionic parasympathetic
nerves relaxes coro-nary arteriolar smooth muscle via the NO/cGMP pathway in
humans as described above. Damage to the endothelium, as occurs with
atherosclerosis, eliminates this action, and acetylcho-line is then able to
contract arterial smooth muscle and produce vasoconstriction. Parasympathetic
nerve stimulation also causes vasodilation in cerebral blood vessels; however,
the effect often appears as a result of NO released either from NANC
(nitrergic) neurons or as a cotransmitter from cholinergic nerves. The relative
contributions of cholinergic and NANC neurons to the vascular effects of
parasympathetic nerve stimulation are not known for most viscera. Skeletal
muscle receives sympathetic cholinergic vasodilator nerves, but the view that
acetylcholine causes vasodila-tion in this vascular bed has not been verified
experimentally. Nitric oxide, rather than acetylcholine, may be released from
these neurons. However, this vascular bed responds to exogenous cho-line esters
because of the presence of M3 receptors on endothelial and smooth
muscle cells.
The
cardiovascular effects of all the choline esters are similar to those of
acetylcholine—the main difference being in their potency and duration of
action. Because of the resistance of methacholine, carbachol, and bethanechol
to acetylcholinesterase, lower doses given intravenously are sufficient to
produce effects similar to those of acetylcholine, and the duration of action
of these syn-thetic choline esters is longer. The cardiovascular effects of
most of the cholinomimetic natural alkaloids and the synthetic analogs are also
generally similar to those of acetylcholine.
Pilocarpine
is an interesting exception to the above statement. If given intravenously (an
experimental exercise), it may produce hypertension after a brief initial
hypotensive response. The longer-lasting hypertensive effect can be traced to
sympathetic ganglionic discharge caused by activation of postganglionic cell
membrane M1 receptors, which close K+ channels and elicit
slow excitatory (depolarizing) postsynaptic potentials. This effect, like the
hypotensive effect, can be blocked by atropine, an antimuscarinic drug.
3. Respiratory system—Muscarinic stimulants contract thesmooth muscle of the bronchial tree. In addition, the glands of the tracheobronchial mucosa are stimulated to secrete. This combina-tion of effects can occasionally cause symptoms, especially in individuals with asthma. The bronchoconstriction caused by muscarinic agonists is eliminated in knockout animals in which the M3 receptor has been mutated.
4. Gastrointestinal tract—Administration of
muscarinic ago-nists, as in parasympathetic nervous system stimulation,
increases the secretory and motor activity of the gut. The salivary and
gas-tric glands are strongly stimulated; the pancreas and small intesti-nal
glands are stimulated less so. Peristaltic activity is increased throughout the
gut, and most sphincters are relaxed. Stimulation of contraction in this organ
system involves depolarization of the smooth muscle cell membrane and increased
calcium influx.
Muscarinic
agonists do not cause contraction of the ileum in mutant mice lacking M2
and M3 receptors. The M3 receptor is required for direct
activation of smooth muscle contraction, whereas the M2 receptor
reduces cAMP formation and relaxation caused by sympathomimetic drugs.
5. Genitourinary tract— Muscarinic agonists
stimulate thedetrusor muscle and relax the trigone and sphincter muscles of the
bladder, thus promoting voiding. The function of M2 and M3
receptors in the urinary bladder appears to be the same as in intes-tinal
smooth muscle. The human uterus is not notably sensitive to muscarinic
agonists.
6. Miscellaneous secretory
glands—Muscarinic
agonistsstimulate secretion by thermoregulatory sweat, lacrimal, and
nasopharyngeal glands.
7. Central nervous system—The central nervous
system con-tains both muscarinic and nicotinic receptors, the brain being
relatively richer in muscarinic sites and the spinal cord containing a
preponderance of nicotinic sites.
All
five muscarinic receptor subtypes have been detected in the central nervous
system. The roles of M1 through M3 have been analyzed by
means of experiments in knockout mice. The M1 subtype is richly
expressed in brain areas involved in cognition. Knockout of M1
receptors was associated with impaired neuronal plasticity in the forebrain,
and pilocarpine did not induce seizures in M1 mutant mice. The
central nervous system effects of the synthetic muscarinic agonist oxotremorine
(tremor, hypothermia,and antinociception) were lacking in mice with
homozygously mutated M2 receptors. Animals lacking M3
receptors, especially those in the hypothalamus, had reduced appetite and
diminished body fat mass.
In
spite of the smaller ratio of nicotinic to muscarinic recep-tors, nicotine and
lobeline (Figure 7–3) have important effects on the brain stem and cortex.
Activation of nicotinic receptors occurs at presynaptic and postsynaptic loci.
Presynaptic nicotinic recep-tors allow acetylcholine and nicotine to regulate
the release of several neurotransmitters (glutamate, serotonin, GABA, dopamine,
and norepinephrine). Acetylcholine regulates norepineph-rine release via α3β4 nicotinic receptors
in the hippocampus and inhibits acetylcholine release from neurons in the
hippocampus and cortex. The α4β2 oligomer is the most abundant nicotinic
receptor in the brain. Chronic exposure to nicotine has a dual effect at
nicotinic receptors: activation (depolarization) followed by desensitization.
The former effect is associated with greater release of dopamine in the
mesolimbic system. This effect is thought to contribute to the mild alerting
action and the addictive property of nicotine absorbed from tobacco. When the β2 sub-units are
deleted in reconstitution experiments, acetylcholine binding is reduced, as is
the release of dopamine. The later desen-sitization of the nicotinic receptor
is accompanied by increased high-affinity agonist binding and an upregulation
of nicotinic binding sites, especially those of the α4β2 oligomer. Sustained desensitization may
contribute to the benefits of nicotine replace-ment therapy in smoking
cessation regimens. In high concentra-tions, nicotine induces tremor, emesis,
and stimulation of the respiratory center. At still higher levels, nicotine
causes convul-sions, which may terminate in fatal coma. The lethal effects on
the central nervous system and the fact that nicotine is readily absorbed form
the basis for the use of nicotine as an insecticide.
8. Peripheral nervous system—Autonomic ganglia areimportant sites of nicotinic synaptic action. The nicotinic agents shown in Figure 7–3 cause marked activation of these nicotinic receptors and initiate action potentials in postganglionic neurons (see Figure 6–8). Nicotine itself has a somewhat greater affinity for neuronal than for skeletal muscle nicotinic receptors. The action is the same on both parasympathetic and sympathetic ganglia. The initial response therefore often resembles simultaneous discharge of both the parasympathetic and the sympathetic nervous systems. In the case of the cardiovascular system, the effects of nicotine are chiefly sympathomimetic. Dramatic hypertension is produced by parenteral injection of nicotine; sympathetic tachycardia may alternate with a bradycardia mediated by vagal discharge. In the gastrointestinal and urinary tracts, the effects are largely parasym-pathomimetic: nausea, vomiting, diarrhea, and voiding of urine are commonly observed. Prolonged exposure may result in depo-larizing blockade of the ganglia.
Neuronal
nicotinic receptors are present on sensory nerve endings—especially afferent
nerves in coronary arteries and the carotid and aortic bodies as well as on the
glomus cells of the lat-ter. Activation of these receptors by nicotinic
stimulants and of muscarinic receptors on glomus cells by muscarinic stimulants
elicits complex medullary responses, including respiratory altera-tions and
vagal discharge.
9. Neuromuscular junction—The nicotinic receptors
on theneuromuscular end plate apparatus are similar but not identical to the
receptors in the autonomic ganglia (Table 7–1). Both types respond to
acetylcholine and nicotine. (However, as noted, the receptors differ in their
structural requirements for nicotinic blocking drugs.) When a nicotinic agonist
is applied directly (by iontophoresis or by intra-arterial injection), an
immediate depolarization of the end plate results, caused by an increase in
permeability to sodium and potassium ions (Figure 7–4). The contractile
response varies from disorganized fascicula-tions of independent motor units to
a strong contraction of the entire muscle depending on the synchronization of
depolariza-tion of end plates throughout the muscle. Depolarizing nicotinic agents
that are not rapidly hydrolyzed (like nicotine itself ) cause rapid development
of depolarization blockade; transmission blockade persists even when the
membrane has repolarized. This latter phase of block is manifested as flaccid
paralysis in the case of skeletal muscle.
Related Topics