Chapter: Clinical Anesthesiology: Anesthetic Management: Anesthesia for Patients with Liver Disease
Anesthesia for Cirrhosis
Cirrhosis
Cirrhosis is a serious and progressive
disease that eventually results in hepatic failure, and the most common cause
of cirrhosis in the United States is chronic alcohol abuse. Other causes
include chronic active hepatitis (postnecrotic cirrhosis), chronic bili-ary
inflammation or obstruction (primary biliary cirrhosis, sclerosing
cholangitis), chronic right-sided congestive heart failure (cardiac cirrhosis),
autoimmune hepatitis, hemochromatosis, Wilson’s disease, α1-antitrypsin
deficiency, nonalcoholic ste-atohepatitis, and cryptogenic cirrhosis.
Regardless of the cause, hepatocyte necrosis is followed by fibrosis and
nodular regeneration. Distortion of the liver’s normal cellular and vascular
architecture obstructs portal venous flow and leads to portal hypertension,
whereas impairment of the liver’s nor-mal synthetic and other diverse metabolic
functions results in multisystem disease. Clinically, signs and symptoms often
do not correlate with disease sever-ity. Manifestations are typically absent
initially, but jaundice and ascites eventually develop in most patients. Other
signs include spider angiomas, pal-mar erythema, gynecomastia, and
splenomegaly. Moreover, cirrhosis is generally associated with the development
of three major complications: (1) vari-ceal hemorrhage from portal
hypertension, intractable fluid retention in the form of ascites and the hepatorenal
syndrome, and (3) hepaticencephalopathy or coma.
Approximately 10% of patients with cirrhosis also develop at least one
episode of spontaneous bacterial peritonitis, and some patients eventually
develop hepatocellular carcinoma.
A few diseases can produce hepatic fibrosis without hepatocellular
necrosis or nodular regener-ation, resulting in portal hypertension and its
asso-ciated complications with hepatocellular function often preserved. These
disorders include schistoso-miasis, idiopathic portal fibrosis (Banti’s
syndrome), and congenital hepatic fibrosis. Obstruction of the hepatic veins or
inferior vena cava (Budd–Chiari syndrome) can also cause portal hypertension.
The latter may be the result of venous thrombosis (hypercoagulable state), a tumor
thrombus (eg, renal carcinoma), or occlusive disease of the sublobular hepatic
veins.
Preoperative Considerations
The detrimental effects of anesthesia and sur-gery on hepatic blood flow
are discussed below. Patients with cirrhosis are at increased risk of
deterioration of liver function because of their limited functional reserves.
Successful anesthetic management of these patients is dependent on recognizing
the multisystem nature of cirrhosis
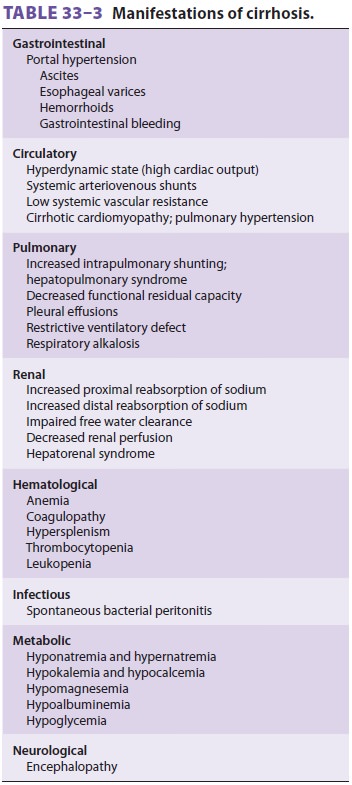
(Table 33–3) and controlling or preventing its
complications.
A. Gastrointestinal Manifestations
Portal hypertension leads to the development of extensive portosystemic
venous collateral channels.
Four major collateral sites are generally
recognized: gastroesophageal, hemorrhoidal, periumbilical, and retroperitoneal.
Portal hypertension is often apparent preoperatively, as evidenced by dilated
abdominalwall veins (caput medusae).
Massive bleeding from gastroesophageal varices is a major causeof morbidity and
mortality, and, in addition to the effects of acute blood loss, the absorbed
nitrogen load from the breakdown of blood in the intestinal tract can
precipitate hepatic encephalopathy.
The treatment of variceal bleeding is primarily
supportive, but frequently involves endoscopic pro-cedures for identification
of the bleeding site(s) and therapeutic maneuvers, such as injection sclerosis
of varices, monopolar and bipolar electrocoagulation, or application of
hemoclips or bands. In addition to the risks posed by a patient who is
physiologi-cally fragile and acutely hypovolemic and hypo-tensive, anesthesia
for such endoscopic procedures frequently involves the additional challenges of
an encephalopathic and uncooperative patient and a stomach full of food and
blood. Endoscopic uni-polar electrocautery may adversely affect implanted
cardiac pacing and defibrillator devices.
Blood loss should be replaced with intra-venous fluids and blood
products. Nonsurgical treatment includes vasopressin, somatostatin,
propranolol, and balloon tamponade with a Sengstaken–Blakemore tube.
Vasopressin, soma-tostatin, and propranolol reduce the rate of blood loss. High
doses of vasopressin can result in con-gestive heart failure or myocardial
ischemia; con-comitant infusion of intravenous nitroglycerin may reduce the
likelihood of these complications and bleeding. Placement of a percutaneous
transjugu-lar intrahepatic portosystemic shunt (TIPS) can reduce portal
hypertension and subsequent bleed-ing, but may increase the incidence of
encepha-lopathy. When the bleeding fails to stop or recurs, emergency surgery
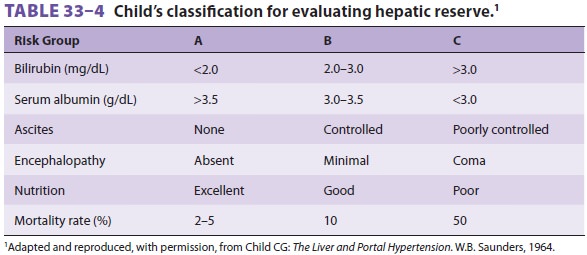
may be indicated. Surgical risk has been shown to correlate with the degree of
hepatic impairment, based on clinical and labora-tory findings. Child’s
classification for evaluating hepatic reserve is shown in Table
33–4. Shunting procedures are generally performed on low-risk patients,
whereas ablative surgery, esophageal tran-section, and gastric
devascularization are reserved for high-risk patients.
B. Hematologic Manifestations
Anemia, thrombocytopenia, and, less commonly,
leukopenia, may be present. The cause of the ane-mia is usually multifactorial
and includes blood loss, increased red blood cell destruction, bone marrow
suppression, and nutritional deficiencies. Congestive splenomegaly secondary to
portal hypertension is largely responsible for the thrombocytopenia and
leukopenia. Coagulation factor deficiencies arise as a result of decreased
hepatic synthesis. Enhanced fibrinolysis secondary to decreased clearance of
acti-vators of the fibrinolytic system may also contribute to the coagulopathy.
The need for preoperative blood transfusions should be balanced against
the obligatory increase in nitrogen load. Protein breakdown from excessive blood
transfusions can precipitate encephalopathy. However, coagulopathy should be
corrected before surgery. Clotting factors should be replaced with appropriate
blood products, such as fresh frozen plasma and cryoprecipitate. Platelet
transfusions should be considered immediately prior to surgery for counts less
than 75,000/µL.
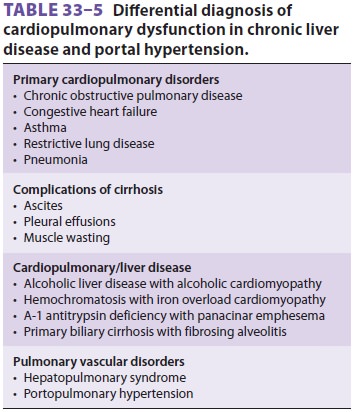
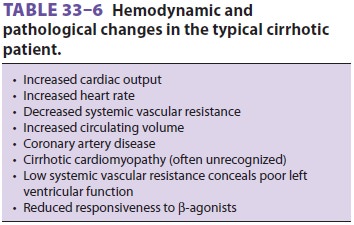
C. Circulatory Manifestations
End-stage liver disease, and, in particular, cirrhosis of the liver, may
be associated with disorders of all major organ systems (Tables
33–3 and 33–5). The cardiovascular changes observed in the patient with hepatic cirrhosis
are usually that of a hyperdynamic circulation, although clinically
signif-icant cirrhotic cardiomyopathy is often present and not recognized (Table
33–6). There may be a reduced cardiac contractile response to stress,
altered dia-stolic relaxation, downregulation of β-adrenergic receptors, and electrophysiological changes as a result of
cirrhotic cardiomyopathy.
Echocardiographic examination of cardiac function may initially be interpreted
as normal because of significant afterload reduction caused by low systemic
vascular resistance. However, both
systolic and diastolic dysfunction are often found. Noninvasive stress
imaging is frequently used to assess coronary artery disease in patients older
than age 50 years and those with risk factors.
Hepatopulmonary Syndrome
The effects of hepatic
cirrhosis on the pulmo-nary vascular resistance (PVR) vessels mayresult in
chronic hypoxemia. Hepatopulmonary
syn-drome (Table 33–7) is found
in approximately 30%of liver transplant candidates and is characterized
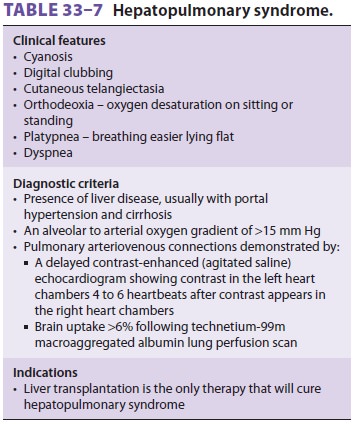
by pulmonary arteriolar endothelial dysfunction. The resultant intrapulmonary
vascular dilata-tion causes intrapulmonary right-to-left shunting and an
increase in the alveolar to arterial oxygen gradient.
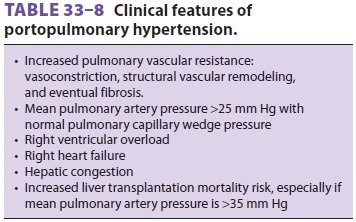
Portopulmonary Hypertension
Pulmonary vascular remodeling may occur in association with chronic
liver disease, involving vascular smooth muscle proliferation,
vasoconstric-tion, intimal proliferation, and eventual fibrosis, all presenting
as an obstructive pathology that causes an increased resistance to flow. This
may result in pulmonary hypertension; if associated with portal hypertension,
it is termed portopulmonary hyperten-sion
(POPH;Table 33–8).
The diagnostic criteria for POPH include a
mean pulmonary artery pressure (mPAP) >25 mm Hg at rest, and a PVR > 240 dyn.s.cm−5.
The transpulmo-nary gradient of >12 mm Hg (mPAP – pulmonary arteriolar occlusion pressure [PAOP])
reflects the obstruction to flow and distinguishes the contribu-tion of volume
and resistance to the increase in mPAP.
POPH may be classified as mild (mPAP 25–35 mm
Hg), moderate (mPAP 35 and 45 mm Hg), and severe (mPAP 45 mm Hg). Mild
POPH is not associated with increased mortality at liver transplantation,
although the immediate recovery period may be challenging if there is a
significant increase in cardiac output after reperfusion of the new graft.
Moderate and severe POPH are associ-ated with significant mortality at
transplantation. However, the key factor is not mPAP, but rather right
ventricular (RV) function.
The success of liver transplantation will
depend on the right ventricle maintaining good function during and after the
transplant procedure despite increases in cardiac output, volume, and PVR. If
RV dysfunction or failure occurs, graft congestion with possible failure and
serious morbidity, includ-ing mortality, may ensue. Assessment of the right
ventricle using transesophageal echocardiography (TEE) is often helpful.
The role of liver transplantation in the
manage-ment of POPH is not well defined. In some patients, pulmonary
hypertension will reverse quickly after transplant; however, other patients may
require months or years of ongoing vasodilator therapy. Other patients may
continue to progress and even-tually develop RV failure. Some patients will
develop pulmonary hypertension after liver transplantation. Liver
transplantation offers the best outcome in patients with POPH that is
responsive to vasodilator therapy.
D. Respiratory Manifestations
Disturbances in pulmonary gas exchange and
venti-latory mechanics are often present. Hyperventilation is common and
results in a primary respiratory alka-losis. As noted above, hypoxemia is
frequently pres-ent and is due to right-to-left shunting of up to 40% of
cardiac output. Shunting is due to an increase in both pulmonary arteriovenous
communications (absolute) and ventilation/perfusion mismatch-ing (relative).
Elevation of the diaphragm from ascites decreases lung volume, particularly
func-tional residual capacity, and predisposes to atelec-tasis. Moreover, large
amounts of ascites produce a restrictive ventilatory defect that increases the
work of breathing.
Review of the chest radiograph and arterial blood gas measurements is
useful preoperatively because atelectasis and hypoxemia are usually not evident
on clinical examination. Paracentesis should be considered in patients with
massive ascites and pulmonary compromise, but should be performed with caution
because excessive fluid removal can lead to circulatory collapse.
E. Renal Manifestations and Fluid Balance
Derangements of fluid and electrolyte balance
may manifest as ascites, edema, electrolyte disturbances , and hepatorenal
syndrome. Important mechanisms responsible for ascites include (1) portal
hyper-tension, which increases hydrostatic pressure and favors transudation of
fluid across the intestine into the peritoneal cavity; (2) hypoalbuminemia,
which decreases plasma oncotic pressure and favors fluid transudation; (3)
seepage of protein-rich lymphatic fluid from the serosal surface of the liver
secondary to distortion and obstruction of lymphatic channels in the liver; and
(4) avid renal sodium and water retention.
Patients with cirrhosis and ascites have
decreased renal perfusion, altered intrarenal hemo-dynamics, enhanced proximal
and distal sodium reabsorption, and often an impairment of free water
clearance. Hyponatremia and hypokalemia are com-mon. The former is dilutional,
whereas the latter is due to excessive urinary potassium losses (from
sec-ondary hyperaldosteronism or diuretics). The most severe expression of
these abnormalities is seen with the development of hepatorenal syndrome.
Patients with ascites have elevated levels of circulating cate-cholamines,
probably due to enhanced sympathetic outflow. In addition to increased renin
and angio-tensin II, these patients are insensitive to circulating atrial
natriuretic peptide.
Hepatorenal
syndrome is a functional renaldefect in patients with
cirrhosis that usually follows
gastrointestinal bleeding, aggressive diuresis, sepsis, or major surgery. It is
characterized by pro-gressive oliguria with avid sodium retention, azo-temia,
intractable ascites, and a very high mortality rate. Treatment is supportive
and often unsuccessful unless liver transplantation is undertaken.
Judicious perioperative fluid management in
patients with advanced liver disease is critical. The importance of preserving
kidney function periop-eratively cannot be overemphasized. Overzealous
preoperative diuresis should be avoided, and acute intravascular fluid deficits
should be corrected with colloid infusions. Diuresis of ascites and edema fluid
should be accomplished over several days. Loop diuretics are administered only
after measures such as bed rest, sodium restriction (<2 g NaCl/d), and spironolactone are deemed
ineffective. Daily body weight measurements are useful in prevent-ing
intravascular volume depletion during diuresis.
In patients with both ascites and peripheral edema, no more than 1
kg/day should be lost during diuresis; in those with ascites alone, no more
than 0.5 kg/day should be lost. Hyponatremia (serum [Na+] < 130 mEq/L) also requires water
restric-tion (<1.5 L/d),
and potassium deficits should be replaced preoperatively.
F. Central Nervous System Manifestations
Hepatic encephalopathy is characterized by
altera-tions in mental status with fluctuating neurological signs (asterixis,
hyperreflexia, and/or inverted plan-tar reflex) and characteristic
electroencephalo-graphic changes (symmetric high-voltage, slow-wave activity).
Some patients also have elevated intracra-nial pressure. Metabolic
encephalopathy seems to be related to both the amount of hepatocellular damage
present and the degree of shunting of portal blood away from the liver and directly
into the systemic circulation. The accumulation of substances origi-nating in
the gastrointestinal tract (but normally metabolized by the liver) has been
implicated
Factors known to precipitate hepatic
encepha-lopathy include gastrointestinal bleeding,increased
dietary protein intake, hypokalemic alka-losis from vomiting or diuresis,
infections, and worsening liver function.
Hepatic encephalopathy should be aggressively treated preoperatively.
Precipitating causes should be corrected. Oral lactulose 30–50 mL every 8 hr or
neomycin 500 mg every 6 hr is useful in reduc-ing intestinal ammonia
absorption. Lactulose acts as an osmotic laxative, and, like neomycin, likely
inhibits ammonia production by intestinal bacteria. Sedatives should be
avoided.
Intraoperative Considerations
Patients with postnecrotic cirrhosis due to hepatitis B or hepatitis C
who are carriers of the virus may be infectious. Universal precautions are
always indi-cated in preventing contact with blood and body flu-ids from all
patients.
A. Drug Responses
The response to anesthetic agents is
unpredictable in patients with cirrhosis. Changes in central nervous system
sensitivity, volumes of distribution, protein binding, drug metabolism, and
drug elimination are common. An increase in the volume of distribution for
highly ionized drugs, such as neuromuscular blockers (NMBs), is due to the
expanded extracel-lular fluid compartment; an apparent resistance may be
observed, requiring larger than normal loading doses. However, smaller than normal
maintenance doses of NMBs dependent on hepatic elimination (pancuronium,
rocuronium, and vecuronium) are needed. The duration of action of
succinylcho-line may be prolonged because of reduced levels of
pseudocholinesterase, but this is rarely of clinical consequence.
B. Anesthetic Technique
The cirrhotic liver is very dependent on
hepatic arterial perfusion because of reduced portal venous blood flow.
Preservation of hepatic arterial blood flow and avoidance of agents with
potentially adverse effects on hepatic function are critical. Regional
anesthesia may be used in patients without throm-bocytopenia or coagulopathy,
but hypotension must be avoided. A propofol induction followed by iso-flurane
or sevoflurane in oxygen or an oxygen–air mixture is commonly employed for
general anesthe-sia. Opioid supplementation reduces the dose of the volatile
agent required, but the half-lives of opioids are often significantly
prolonged, which may cause prolonged postoperative respiratory depression.
Cisatracurium may be the NMB of choice because of its nonhepatic metabolism.
Preoperative nausea, vomiting, upper gastroin-testinal bleeding, and
abdominal distention due to massive ascites require a well-planned anesthetic
induction. Preoxygenation and a rapid-sequence induction with cricoid pressure
are often performed. In unstable patients and those with active bleeding,
either an awake intubation or a rapid-sequence induction using ketamine or
etomidate and succi-nylcholine is suggested.
C. Monitoring
Pulse oximetry should be supplemented with
arterial blood gas measurements to monitor acid–base sta-tus. Patients with
large right-to-left intrapulmonary shunts may not tolerate the addition of
nitrous oxide and may require positive end-expiratory pressure (PEEP) to treat
ventilation/perfusion inequalities and subsequent hypoxemia. Patients receiving
vaso-pressin infusions should be monitored for myocar-dial ischemia from
coronary vasoconstriction.
Continuous intraarterial pressure monitoring is often used because hemodynamic
instability fre-quently occurs as a result of excessive bleeding and surgical
manipulations. Intravascular volume sta-tus is often difficult to optimize, and
goal-directed hemodynamic and fluid therapy utilizing esopha-geal Doppler,
arterial waveform analysis, or echocar-diography should be considered. Such
approaches may be helpful in preventing the hepatorenal syn-drome. Urinary
output must be followed closely; mannitol may be considered for persistently
low urinary outputs despite adequate intravascular fluid replacement.
D. Fluid Replacement
Most patients are sodium-restricted
preoperatively, but preservation of intravascular volume and uri-nary output
takes priority intraoperatively. The use of predominantly colloid intravenous
fluids (albumin) may be preferable to avoid sodium over-load and to increase
oncotic pressure. Intravenous fluid replacement should take into account the
excessive bleeding and fluid shifts that often occur in these patients during
abdominal procedures. Venous engorgement from portal hypertension, lysis of
adhesions from previous surgery, and coag-ulopathy lead to excessive bleeding
during surgical procedures, whereas evacuation of ascites and pro-longed
surgical procedures result in large fluidshifts. Following the removal of large
amounts of ascitic fluid, aggressive intravenous fluidreplacement is often
necessary to prevent profound hypotension and kidney failure.
Most preoperative patients are anemic and
coag-ulopathic, and perioperative red blood cell transfusion may lead to
hypocalcemia (citrate toxicity) because of elevated plasma citrate levels
resulting from impaired citrate metabolism in the cirrhotic liver. Citrate, the
anticoagulant in stored red blood cell preparations, binds with plasma calcium,
producing hypocalcemia. Intravenous calcium is often necessary to reverse the
negative inotropic effects of decreased blood ionized calcium concentration .
Related Topics