Chapter: Biochemical Pharmacology : Pharmacology of Eicosanoids
Cyclooxygenase inhibitors
Cyclooxygenase inhibitors
Cyclooxygenase occurs in three isoforms in the
mammalian organism:
• Cox-1 is constitutively expressed and
responsible for most of the `housekeeping' functions of eicosanoids, including
processes such as calcium metabolism in the bone, and stomach mucous membrane
maintenance. It is also responsible for synthesis of thromboxanes in
thrombocytes and of prostacyclin (PGI) in endothelial cells, which have antagonistic
function in thrombocyte aggregation and activation (see later).
• Cox-2 is inducible and mostly expressed in
inflammato-ry cells; it is considered the main culprit in the release of
prostaglandins at sites of inflammation. Since anti-in-flammatory therapy is
the main therapeutic application of Cox inhibitors, there is considerable
interest in the development of drugs selectively acting on this form.
• Cox-3 is a splice variant of Cox-1. It has been
characterized in the brain of dogs. Its selective inhibition by ac-etaminophen,
along with the antipyretic and analgesic5 activity of that drug,
suggests a major role of Cox-3 in triggering fever and pain. However, if the
homologous splice sites were used with a primary transcript of the human Cox-1
gene, this would give rise to an mRNA containing a premature stop codon. It is
not clear at present whether humans actually do possess a third vari-ant of Cox
at all.
Most of
the drugs that inhibit cyclooxygenase do inhib-it both Cox-1 and Cox-2; this
applies to drugs such as di-clofenac, indomethacine, and acetylsalicylic acid.
More re-cent developments have led to selective inhibitors of Cox Crystal
structures of inhibitors bound to Cox-1 and Cox-2have been obtained, and they
can be used together with mutagenesis experiments to understand the molecular
in-teractions of inhibitor molecules with the active site. Such knowledge is
useful, since it allows the development of more selective or more effective
inhibitors to proceed in a targeted way.
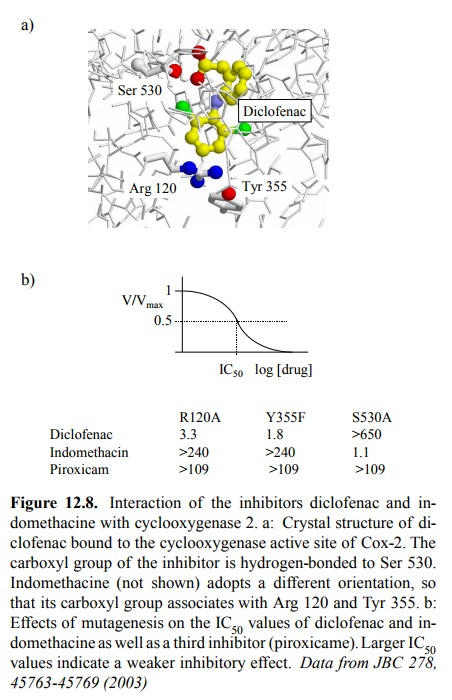
As an
example, the binding of diclofenac to the active site of Cox-2 is shown; its
carboxyl group binds to the serine 530 residue (Figure 12.8a). In contrast, the
carboxyl group of indomethacin points to arginine 120 and Tyr 355, much in the
same way as the carboxylate of arachidonic acid does (not shown). Results from
mutagenesis experiments cor-respond well with these findings (Figure 12.8b):
While re placement of arginine 120 or tyrosine 355 strongly reduces the
inhibitory potency of indomethacin but not diclofenac, the opposite behaviour
is observed with the removal of ser-ine 530. All three residues are important
with a third in-hibitor (piroxicam; Figure 12.9).
While diclofenac and most
other cyclooxygenase inhibitors act competitively (i.e., non-covalently),
acetylsalicylic acid causes covalent modification of serine 530. Its effect may
therefore last longer than that of a non-covalent inhibitor. Interestingly, the
half-life of acetylsalicylic acid is rather short – about 15 minutes; most of
the drug is just hydrol-ysed to acetic acid and salicylic acid. However,
salicylic acid itself still acts as a (competitive) inhibitor of Cox. Also, the
covalent modification of Cox achieved early on will persist after elimination
of acetylsalicylic acid, so that the clinical effect of this drug will outlast
its elimination.
The
covalent, irreversible mode of action of acetylsali-cylic acid is important in
its use for inhibiting thrombocyte aggregation in patients with cardiovascular
disease (more specifically, atherosclerosis). This is a practically very im
portant application, since atherosclerosis is a very common disease,
particularly in western countries.
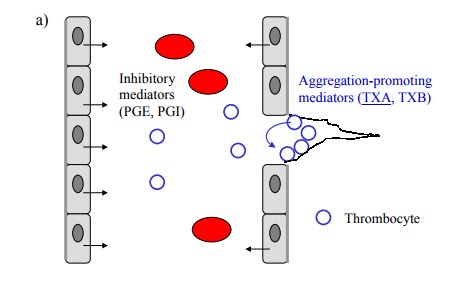
Thrombocyte aggregation is
promoted by thromboxanes, which are synthesized in thrombocytes, and is
inhibited by prostaglandins I and E, which are released by endothelial cells
(Figure 12.10a). As all of these are derived via Cox-1, we need to selectively
inhibit Cox 1 in thrombocytes but not endothelial cells. How can such
selectivity be possibly achieved? The solution to this dilemma lies in the
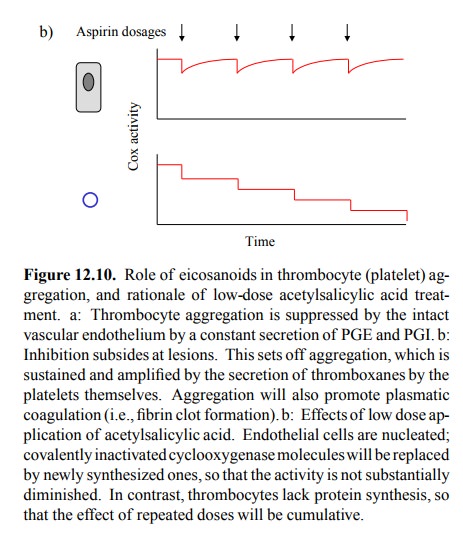
different lifetimes of Cox-1 in the two cell types: In endothelial cells, the
enzyme is turned over within hours; inactivated enzyme molecules will thus be
replaced by newly synthesized ones. Thrombocytes, however, don't have a nucleus
and there-fore lack protein synthesis; irreversibly inactivated enzyme
molecules will therefore never be replaced (Figure 12.10b). If we properly
adjust the dosage of acetylsalicylic acid, we can indeed maintain the enzyme
activity in the endothelium yet efficiently inhibit it in the thrombocytes.
Increasing the dosage, indeed, will reduce the beneficial drug effect, since
both PGI/PGE and TXA synthesis will now be inhibited.
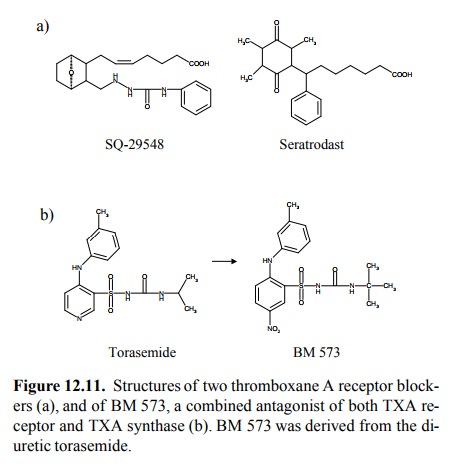
Another,
more recent principle of limiting thrombocyte aggregation consists in the use
of thromboxane receptor blockers. Examples are shown in Figure 12.11a. There
even are drugs (presently experimental) that potently inhib-it both the
thromboxane receptor and the enzyme throm-boxane A synthase. It is rather
intriguing that one such drug molecule (BM-573, Figure 12.11b) could be
obtained by only slight modification of a precursor (torasemide) that has only
weak inhibitory activity at the receptor, and lacks any obvious similarity with
thromboxane altogether.
Related Topics