Chapter: Medical Immunology: Malignancies of the Immune System
Acute Lymphocytic Leukemias
Acute Lymphocytic Leukemias
Acute lymphocytic leukemias are those acute leukemias in which the malignant cells seen in the peripheral blood are immature lymphocytes (lymphoblasts). These leukemias usually have a very poor prognosis. Death usually occurs as a consequence of the massive lym-phocytic proliferation in the bone marrow, where the proliferating cells overwhelm and smother the normal hematopoietic cells.
1. Classification
With the introduction of monoclonal antibodies directed against T- and B-cell markers, it was determined that the large majority (about 95%) of acute lymphocytic leukemia are B-cell–derived because the proliferating cells express the CD19 and CD20 B cell markers. The remaining 5% of these leukemias are of the T-cell type.
2. Enzymatic Markers of Acute Lymphocytic Leukemia
The expression of enzymes of the purine salvage pathway is altered in acute lymphocytic leukemia. Adenosine deaminase (ADA) is often overexpressed. In patients with increased ADA, 2-deoxycorfomycin, a drug that specifically inhibits ADA, has remarkable thera-peutic effects. Terminal deoxynucleotidyl transferase (Tdt) is not expressed by adult lym-phocytes but is reexpressed by about 80% of all cases of this type of leukemia; it consti-tutes a useful marker because its levels fall during remission and increase again before a clinically apparent relapse.
3. B-Cell Acute Lymphocytic Leukemia
In most cases the malignant cells do not express membrane immunoglobulins, do not have intracellular immunoglobulins, have rearranged heavy-chain genes, and express the com-mon acute lymphocytic leukemia antigen (CALLA). CALLA is present in the majority of non-T, non-B acute lymphocytic leukemia lymphocytes, almost always expressed in asso-ciation with a B-cell marker such as the CD19 or CD20 antigen. CALLA is also expressed by the lymphoblasts seen during the blastic crisis of patients with chronic myelocytic leukemia (CML); these lymphoblasts also express B-cell markers such as the CD20 anti-gen, establishing their identity as B-cell precursors. When the blast cells seen in the blastic crisis of a patient with CML are CALLA+ and CD20+ , the crisis responds well to chemotherapy; when none of these markers is expressed, survival is limited to a few days. In acute lymphocytic leukemia CALLA positivity identifies patients with a more favorable prognosis. On the other hand, acute lymphocytic leukemia in which the leukemic cells ex-press membrane or cytoplasmic immunoglobulins has very poor prognosis, and patients survive less than a year unless very aggressively treated.
Monoclonal anti-CALLA antibodies have been used therapeutically in acute lym-phocytic leukemia with disappointing results. A sharp decrease in leukemic cell counts is observed after administration of antibody, but this effect is usually of short duration since the CALLA+ lymphocytic population is soon replaced by a CALLA- population (anti-genic modulation) not affected by further administration of antibody. Also, the prolonged administration of monoclonal anti-CALLA of murine origin leads to the development of antimouse immunoglobulin antibodies, which cause rapid elimination of anti-CALLA an-tibodies from the patient’s circulation and may also cause serum sickness.
4. T-Cell Acute Lymphocytic Leukemia
This type of leukemia usually has a worse prognosis than B-cell acute lymphocytic leukemia (patients with T-cell acute lymphocytic leukemia have a less than 20% probabil-ity of remaining in remission for more than 2 years). Chromosomal abnormalities involv-ing the T-cell receptor genes have been observed in at least 40% of the T-cell leukemias. One of the most frequent is a translocation of the area of chromosome 14, which carries the α gene of the T-cell receptor to the area of chromosome 8, which has the c-myc gene. Equally frequent is a translocation of the area of chromosome 7 that contains the βchain of the T cell receptor to chromosome.
Three distinct subgroups of leukemic processes can be defined in the group of T-cell acute lymphocytic leukemia depending on the expression of T-cell membrane markers (Table 27.3):
1. The first and largest group includes cases in which the proliferating T cells ex-press the CD5 and CD2 markers. The malignant cell is, therefore, an early T-cell precursor that mutated before the full rearrangement of the T-cell receptor genes so that the CD3 molecule is not expressed.
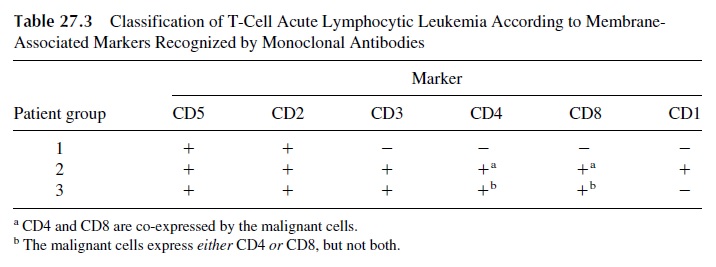
2. The second group is constituted by cases in which the proliferating cells have reached a later stage of T-cell differentiation. The malignant cells express CD3, co-express both CD4 and CD8 markers, and are also positive for the CD1 marker, indicating an aberrant reversal of a partially differentiated T lymphocyte to an earlier ontogenic stage.
3. The third group is constituted by cases with proliferating mature T cells, sharing markers (CD4 or CD8, in association with both CD2 and CD3) with the lym-phocytes normally found in the peripheral blood and lymphoid organs.
Because of the relative rarity of T-cell acute lymphocytic leukemia, it has not yet been possible to establish whether any of the subgroups of this disease has a worse prog-nosis than that of T-cell acute lymphocytic leukemia in general.
5. T-Cell Leukemia Associated to HTLV-I
This type of T-cell leukemia has a very unique geographic distribution, closely associated to the first identified human retrovirus (Human T-cell lymphotropic virus-I, or HTLV-I), which is very prevalent in Japan and the Caribbean basin (where the rates of infection reach endemic proportions); the virus has also been reported, although with lower frequency, in the southern United States.
HTLV-I–associated T-cell leukemia develops 10–20 years after infection with the virus. This very long latency period and the fact that T-acute lymphocytic leukemia is only seen in a fraction of the HTLV-I–infected individuals (4–5% of the seropositive individu-als) suggests that malignant transformation does not result exclusively from the viral in-fection. However, the nature of the additional cofactors leading to leukemic transformation is unknown.
HTLV-I is an exogenous retrovirus, fully able to replicate and to be transmitted hor-izontally. Its genome contains a transforming gene, tax, whose gene product modifies the nuclear binding protein NFkB, leading to the permanent overexpression of IL-2 receptors (CD25) in the infected cells. This has several consequences:
1. This type of T-cell leukemia is easily distinguishable by the fact that the prolif-erating cells are easily labeled with anti-CD25 monoclonal antibodies.
2. IL-2 stimulates the growth of the leukemic T cells in long-term culture.
3. Some patients have malignant T cells that not only express CD25 but also release high concentrations of IL-2, leading to an autocrine circuit of T-cell prolifera-tion.
4. Other interleukin-coding genes are also activated, including the one coding for IL-1β , which directly or indirectly causes osteoclast activation. As a conse-quence of osteoclast activation, bone resorption and hypercalcemia are promi-nent in these patients.
Secondary immunosuppression may develop in patients with HTLV-I leukemia. Sev-eral factors contribute to the state of immunosuppression. IL-2 receptors are shed from the membrane of the leukemic cells and can be detected in high concentrations in the circula-tion. Soluble IL-2 receptors diffuse into the extracellular spaces and adsorb the IL-2 that is necessary for the activation of normal T cells, causing a functional deficiency of this inter-leukin. In addition, The proliferating CD4+ cells function as suppressor-inducers and turn on cells with suppressor activity.
Clinical presentations include skin and bone changes. On the skin, erythroderma and skin ulceration are the most common manifestations and are associated with a dense lym-phocytic infiltration of the dermis and epidermis. It is believed that increased venous per-meability, probably caused by an increased local concentration of IL-2 and other inter-leukins, are responsible for the formation of cellular infiltrates, which, in turn, interfere with proper oxygenation of tissues, leading to localized ischemia and necrosis. Osteoclast activation causes excessive bone resorbtion leading to hypercalcemia, and spontaneous fractures may develop. Reflecting the sate of immunodeficiency common in these patients, opportunistic infections involving organisms such as Pneumocystis carinii pneumonia are relatively frequent.
6. Other HTLV-I–Related Lymphomas
Sézary Syndrome and mycosis fungoides are cutaneous T-cell lymphomas that have also been related to HTLV-I infection.
Sézary syndrome is an exfoliative erythroderma with generalized lymphadenopathy and circulating atypical cells with a characteristic multilobulated nucleus (Sézary cells). The skin is the original site of malignant cell proliferation, and the phase of cutaneous lym-phoma can last many years with little evidence of extracutaneous dissemination. The leukemic evolution is associated with the invasion of the peripheral by malignant cells. The malignant cells infiltrating the skin or circulating in the blood are CD4+ and behave func-tionally as helper T cells when mixed in vitro with T-cell–depleted lymphocytes from a normal donor and antigenically stimulated.
Mycosis fungoides is clinically similar to the cutaneous phase of the Sézary syn-drome, and the infiltrating cells in the skin are also CD4+ . No leukemic stage seems to de-velop in patients afflicted with the disease. However, the lymphocytes from patients with mycosis fungoides suppress the response of normal allogeneic T and B cells.
7. Epstein-Barr Virus–Associated B-Cell Lymphomas
Burkitt’s Lymphoma. Burkitt’s lymphoma (BL), endemic in certain areas ofAfrica and sporadic in the United States, has been characterized as a B-cell lymphoma ex-pressing monotypic surface IgM. Burkitt’s lymphoma is epidemiologically linked to infec-tion of the B lymphocytes with the Epstein-Barr virus (EBV). The malignant B cells in BL usually express a single EBV gene product, the nuclear antigen EBNA-1, which is essen-tial for establishment of latency, but has no known transforming properties. It is possible that the EBV infection has as its main role promoting a state of active B-cell proliferation that may favor the occurrence of the translocations involving the region of chromosome 8 coding for c-myc.
B-Cell Lymphomas in Immunocompromised Patients.B-cell lymphomas arefrequently detected in immunodeficient or iatrogenically immunosuppressed patients, and in almost all cases there is evidence of association with EBV. In those cases, a variety of viral-coded proteins are expressed on the malignant cells, including six different nuclear antigens and three different membrane proteins. Of the proteins coded by nuclear antigens, EBNA-2 protein has immortalizing properties, transactivating the cyclin-2 gene and oth-ers, EBNA-LP impairs the function of the products of two tumor-suppressor genes, p53 and the retinoblastoma gene product, and the latent membrane protein 1 (LMP-1) is considered as a transforming gene whose activity seems to be mediated by the activation of a Ca2+ /calmodulin-dependent protein kinase.
Hodgkin’s Disease. EBV genomes and gene products can be detected in a signifi-cant number of Hodgkin’s disease lymph node biopsies. More significant is the fact that LMP-1 is among the expressed proteins.
Case 27.2 Revisited
The presentation of a native of the Caribbean basin with an erythemato-ulcerative skin rash, pneumonia, and leukocytosis with a marked increased of the CD4 population and of double staining CD4-CD25 lymphocytes, with osteoporosis and hypercalcemia, is typical for an HTLV-I–associated T-cell leukemia.
The most informative test from the diagnostic point of view would be a serological assay for anti-HTLV-I antibodies, which was positive in this patient. In addition, biopsies of the ulcerative skin lesions and enlarged lymph nodes were compatible with lymphoma. The cells infiltrating the biopsed lymph node were identified as CD4+ , CD25+ . Peripheral blood lymphocytes showed a vigorous mitogenic response to IL-2, circulating levels of sol-uble IL-2 receptors were high (1000 IU/mL, normal <277 U/mL), and the blood levels of paratohormone were normal.
The skin rash was due to a dense lymphocytic infiltration of the dermis and epider-mis, secondary to increased venous permeability, probably caused by elevated local con-centrations of IL-2 and other interleukins. The intense cellular infiltrate interferes with proper oxygenation of tissues, and the resulting ischemia leads to localized necrosis.
Transformed CD4+ cells produce IL-1β and other less well-defined mediators that activate osteoclasts, and induce bone resorption and hypercalcemia. Because of the loss of calcium the bones become fragile and may break with minimal trauma.
ATLL is a malignancy of mature CD3+ , CD4+ T cells caused by the HTLV-I virus. The virus has a transforming gene (tax), which becomes overexpressed, and the protein coded by that gene modifies cellular transactivating proteins, such as NFkB, increasing their activity. As a consequence, the transformed cells overexpress IL-2 receptors and are able to proliferate spontaneously by using their own IL-2 (whose synthesis is also enhanced by the tax gene product) as a growth factor.
The bilateral interstitial infiltrates suggest a pneumonitic process, which could be due to viruses or fungi, such as Pneumocystis carinii. P. carinii is one of the most preva-lent causes of opportunistic pneumonia in immunocompromised individuals. This patient was immunocompromised, as shown by his very low levels of circulating immunoglobulins. In addition, lymphocyte mitogenic responses to PHA and ConA were depressed, revealing a functional impairment of cell-mediated immunity. Examination of a broncho-alveolar lavage sample was positive for the typical silver-staining cysts of P. carinii.
Related Topics