Chapter: Medical Immunology: Malignancies of the Immune System
Physiopathology of B-Cell Dyscrasias
Physiopathology of B-Cell Dyscrasias
1. Direct Pathological Consequences of Malignant B-Cell Proliferation
Depending on the type of proliferating B cell, patients may present:
Enlargement of lymph nodes, spleen, and liver, as seen in lymphomas and some leukemias.
Leukemic invasion of peripheral blood, characteristic of B-cell leukemias.Compressive and obstructive symptoms resulting from the proliferation of plasma cells in soft tissues. Oro-pharyngeal plasmocytomas often lead to obstructive symptoms. Heavy-chain–producing intestinal lymphomas, when grossly nodu-lar, can lead to intestinal obstruction.
Intestinal malabsorption, typical of α-chain disease, resulting from extensive infil-tration of the intestinal submucosa by malignant B cells, causing total disrup-tion of the normal submucosal architecture.
2. General Metabolic Disturbances
General metabolic disturbances are responsible for some major pathological manifestations of B-cell dyscrasia, such as bone destruction, renal insufficiency, anemia, and secondary immunodeficiency.
Bone destruction does not result directly from B-cell proliferation but rather from os-teoclast hyperactivity induced by an activating cascade triggered by the malignant B cells. The initial event is believed to be the exaggerated synthesis of IL-1 and/or lymphotoxin-by transformed plasma cells. These cytokines activate osteoblasts and/or stromal cells and induce the secretion of IL-6, which acts as a growth factor for plasma cells and as an acti-vating factor for osteoclasts. However, there is experimental evidence suggesting that other cytokines, including IL-1 and macrophage colony-stimulating factor (M-CSF), both pro-duced by neoplastic plasma cells, also have activating effects on osteoclasts.
Renal insufficiency can result from a diversity of factors, such as hypercalcemia (sec-ondary to bone reabsorption, hyperuricemia, deposition of amyloid substance in the kidney, clogging of glomeruli or tubuli with paraprotein (favored by dehydration), and plasmocytic infiltration of the kidney.
Anemia (normochromic, normocytic) is frequent and is basically due to decreased production of red cells. A moderate shortening of red cell survival is also common.
A paradoxical feature of B-cell malignancies is the secondary immunodeficiency that develops in patients that often have marked increases in their concentrations of circulating immunoglobulins. This is particularly obvious in patients with of multiple myeloma, who have an increased tendency for pyogenic infections. In reality, if the levels of residual nor-mal immunoglobulins are measured, they are found to be low. Also, these patients show decreased antibody production after active immunization. The depression of the immune response in patients with multiple myeloma appears to be multifactorial.
1. In IgG myeloma, the large amounts of IgG secreted by the malignant cells are likely to have a feedback effect, depressing normal IgG synthesis .
2. A more general mechanism of suppression of the humoral response seems to be mediated by phagocytic monocytes (and to a lesser extent T cells). The im-munosuppressor properties of these cells can be demonstrated by co-culturing peripheral blood mononuclear leukocytes from normal donors and from myeloma patients. The co-culture results in impairment of the function of the normal B lymphocytes.
3. Other abnormalities that may contribute to the predisposition to infections are defects in neutrophil responses and impairment of Fcγ receptor functions in phagocytes, which are more likely to be present in patients with renal failure. Anemia also seems to predispose to infections, for unknown reasons.
In chronic lymphocytic leukemia, in addition to a depression of humoral immunity (milder than that seen in multiple myeloma), there is a depression of T-cell counts and func-tion. Viral and fungal infections, as well as cases of disseminated infection after adminis-tration of live attenuated viral vaccines, have been reported in patients with this type of leukemia.
3. Serum Hyperviscosity
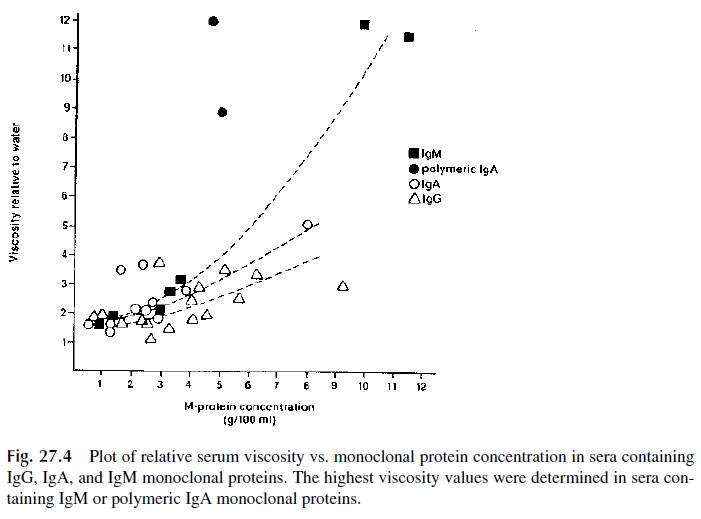
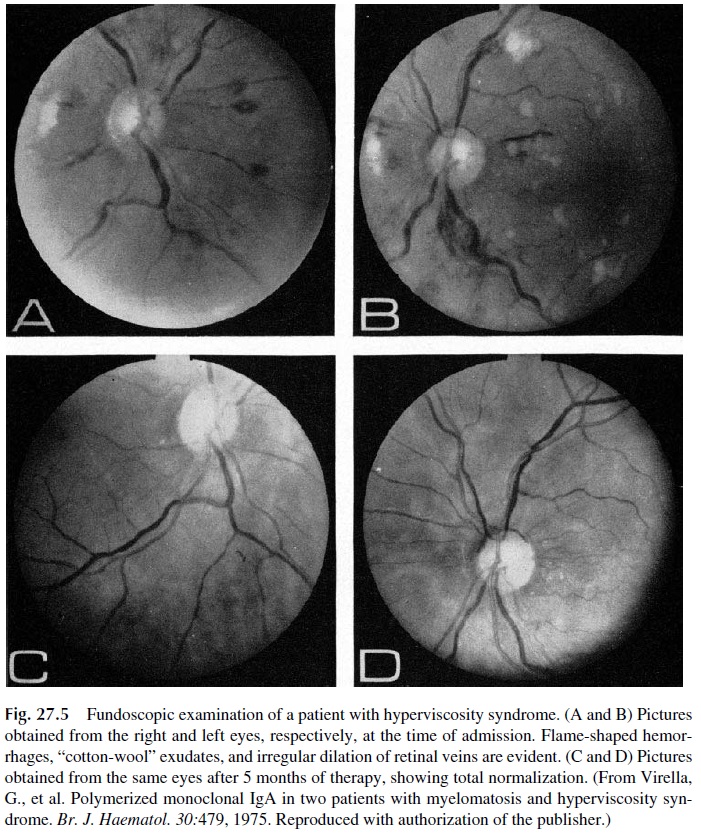
Some plasma cell dyscrasias may present with a constellation of symptoms known as the hyperviscosity syndrome. This is a direct result of serum hyperviscosity, caused by high concentrations of monoclonal proteins. Serum viscosity increases with protein concentra-tion. IgM and polymeric IgA, due to their molecular complexity and high intrinsic viscos-ity, lead to disproportionate increases of blood viscosity (Fig. 27.4). Not surprisingly, the hyperviscosity syndrome is a frequent manifestation of Waldenström’s macroglobuline-mia, a B-cell dyscrasia defined by the synthesis of monoclonal IgM. However, the hyper-viscosity syndrome is also observed in multiple myeloma patients, mainly in those with IgA paraproteins, and occasionally in IgG myeloma, when the concentrations of IgG are very high. The symptoms of serum hyperviscosity are related to high protein concentration, expanded plasma volume, and sluggishness of circulation. Table 27.1 lists the main signs and symptoms of the syndrome. Typical fundoscopic changes are shown in Figure 27.5.
4. Pathological Consequences of the Immunological Activity of a Paraprotein
Most paraproteins have unknown and inconsequential antibody activities, but in some ex-ceptional cases the reactivity of a monoclonal protein may be directly responsible for some of the manifestations of the disease.
Cold Agglutinin Disease.Cold agglutinin disease results from the synthesis oflarge concentrations of monoclonal IgM (IgMk in more than 90% of the cases) with cold agglutinating properties. Those monoclonal cold agglutinins react with the “I” antigen ex-pressed by the erythrocytes of all normal adults, and the reactivity is only evident at tem-peratures below normal body temperature. Usually, the sera suspected to contain them are tested at 4°C on a direct hemagglutination test using O-positive red cells as antigens. For these reasons, these autoantibodies are known as cold agglutinins. Monoclonal cold agglu-tinins can be detected:
1. In cases of IgM-producing B-cell malignancy (Waldenström’s macroglobuline-mia, see below), when the IgM paraprotein behaves as a cold agglutinin. The titers of cold agglutinins in such cases are very high, and usually the patient will have symptoms attributable to the cold agglutinin.
2. In patients with symptomatic cold agglutinin disease associated to high titers of a monoclonal cold agglutinin in which there is no evidence of B-cell dyscrasia other than the presence of a monoclonal anti-I antibody and an increase in the numbers of lymphoplasmocytic cells in the bone marrow.
The clinical manifestations of cold agglutinin disease fall into two categories:
1. Cold-induced hemolytic anemia, which is usually mild, but in some severe cases can be intense enough to lead to acute renal failure
2. Cold-induced ischemia, due to massive intracapillary agglutination in cold-exposed areas
Hyperlipidemia.A pronounced increase in serum lipid and lipoprotein levels canoften be detected in patients with monoclonal gammopathies, and in some cases the mon-oclonal protein has antibody activity to lipoproteins. It has been demonstrated that the bind-ing of antibodies to the lipoprotein molecules alters the uptake and intracellular processing of the lipoprotein, resulting in hyperlipidemia and in increased accumulation of cholesterol in macrophages.
Related Topics