Chapter: Medical Immunology: Malignancies of the Immune System
Clinical Presentations of B-Cell Dyscrasias
Clinical Presentations of B-Cell Dyscrasias
1. Multiple Myeloma
The most frequent clinical symptoms of multiple myeloma are (1) bone pain and “sponta-neous” or “pathological” fractures, (2) malaise, headaches, or other symptoms related to hyperviscosity, (3) weakness and anemia, (4) repeated infections, and (5) renal failure.
In some cases, anemia is the leading feature, and the diagnosis is established when the cause of anemia is investigated. Hemoglobin levels below 7.5 g/dL are usually associ-ated with poor prognosis. Other cases are first seen in a rheumatology outpatient clinic, due to “bone pains.” If there is advanced bone destruction the diagnosis may be prompted by a fracture after minimal trauma (known as “pathological” fractures). Symptoms related to hyperviscosity (see Table 27.1) may also lead to hospitalization.
Repeated infections and renal failure, which usually occur in advanced stages of the disease, are among the most frequent causes of death but rarely constitute the presenting symptoms. Infection is associated with an increased risk of death, and the prognosis for a multiple myeloma patient with renal failure, particularly when his blood urea nitrogen ex-ceeds 80 mg/dL, is also poor.
Several parameters have been found to be associated with rapidly growing myelomas and poor prognosis. In general, these parameters reflect the magnitude of the malignant cell population and its degree of dedifferentiation and include:
· Heavy plasma cell infiltration of the bone marrow ( >30%) Numerous lytic bone lesions
· Bence Jones proteinuria Low hemoglobin ( <11g/L)
· Expression of myeloid cell markers and the CALLA antigen on the membrane of the malignant plasma cells
· Expression of the c-myc gene
· High serum levels of β2-microglobulin, thymidine kinase, and neopterin
Laboratory Diagnosis.A typical case of multiple myeloma will present at leasttwo elements of the following diagnostic triad: (1) bone lesions, (2) monoclonal protein in serum and/or urine, and (3) bone marrow plasmacytosis.
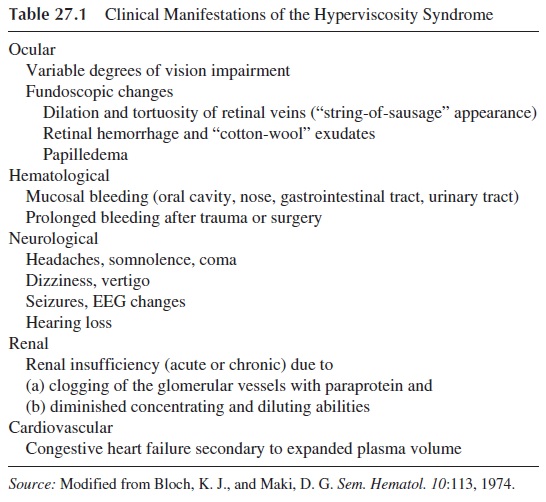
Bone lesions are typically osteolytic (appear in the x-ray as punched-out areas with-out peripheral osteosclerosis) and multiple (several punched-out areas appear in the same bone and can be seen in a number of bones in the same patient) (Fig. 27.6). Practically all bones can be affected. In advanced cases, pathological fractures can occur in the long bones, skull, or spinal column. Rarely, a single bone lesion may be detected in one patient; however, such a “solitary bone plasmacytoma” is in fact rarely solitary, and bone marrow aspiration will reveal diffuse plasmacytosis in most cases. Exceptionally, a patient with monoclonal gammopathy and diffuse plasmacytosis can present with no evident bone le-sions or with generalized osteoporosis.
A monoclonal protein in serum and/or urine can be detected in 98% of the cases of multiple myeloma if proper techniques are used. The distribution of monoclonal proteins among the different immunoglobulin classes closely parallels the relative proportions of those immunoglobulins in normal serum: 60 –70% of the proteins are typed as IgG, 20 –30% as IgA, 1–2% as IgD, and, very rarely, one monoclonal protein can be typed as IgE. A single light-chain type is found in these paraproteins. For example, IgG paraproteins can be either k or λ. The finding of a heterogeneous increase in IgG or any other im-munoglobulin (i.e., and increase of both IgGk and IgGλ molecules) is not compatible with a diagnosis of multiple myeloma.
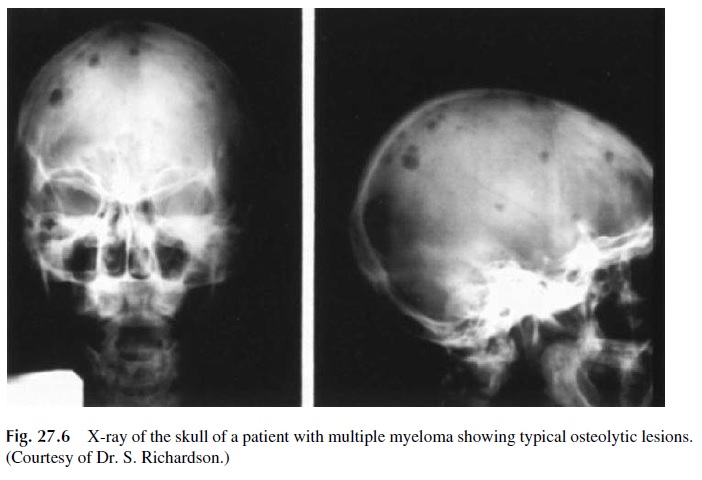
In the urine, the most frequent finding is the elimination of free light chains, k or λ (Bence-Jones proteins) (see Fig. 27.7). These light chains are usually found in addition to a monoclonal immunoglobulin detectable in serum, but in about 20 –30% of patients with multiple myeloma the only abnormal proteins to be found are the free monoclonal light chains in the urine. Some authors give the designation of light-chain disease to the form of multiple myeloma in which the paraprotein consists of free light chains.
Very rarely (about 2% of cases) no monoclonal paraprotein is detected in the serum or urine of a patient with a typical clinical picture or multiple myeloma. This situation is designated as nonsecretory myeloma. In most nonsecretory myelomas, immunofluores-cence studies have demonstrated intracellular monoclonal proteins that are not secreted into the extracellular spaces. Nonsecretory myelomas have a very poor prognosis.
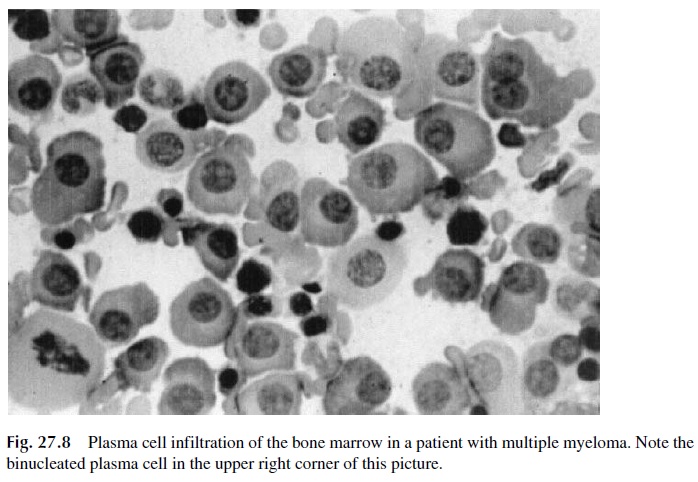
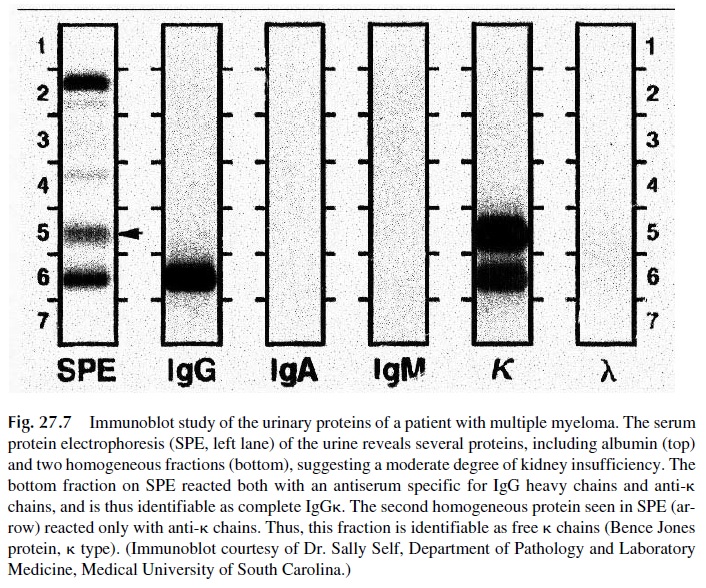
Bone marrow plasmacytosis is the third element of the diagnostic triad for multiple myeloma. Bone marrow aspirates show increased numbers of plasma cells with a more or less mature appearance (Fig. 27.8). The plasma cell infiltration can be massive, with sheets of plasma cells occupying the bone marrow. However, it must be stressed that an increase in the number of plasma cells in the bone marrow, even when associated with morpholog-ical aberrations, is not sufficient to differentiate between malignant and reactive plasma cell proliferation. The differential diagnosis between malignant and reactive plasma cell prolif-eration should be based on the immunochemical characteristics of the patient’s im-munoglobulins. While a patient with a malignant B-cell dyscrasia will show either a mon-oclonal protein or low immunoglobulin levels (if it is a case of nonsecretory myeloma), reactive plasmacytosis is invariably associated with a polyclonal increase of immunoglobulins.
Management.The management of a patient with a monoclonal gammopathy restson a decision about whether the malignancy is dormant or aggressive. Multiple myeloma is, in the vast majority of cases, an aggressive malignancy, and its management is usually based on staging. Staging depends on a variety of parameters indicating early stages versus full-blown or terminal stages, including:
1. A high labeling index, usually determined by counting the number of plasma cells that show bright nuclear staining with a fluorescent monoclonal antibody to 5-bromo-2-deoxyuridine. This enzyme becomes associated with the DNA of ac-tively proliferating cells, and its visualization is considered indicative of an ag-gressive tumor.
2. Other markers associated with aggressive tumors include high levels of (a) β2-microglobulin—produced and shed by actively proliferating plasma cells, (b) thymidine kinase—another enzyme involved in DNA synthesis which is re-leased by dividing cells, (c) IL-6, the main cytokine proposed to mediate plas-mocytoma cell growth, and (d) C-reactive protein—a direct reflection of the lev-els of IL-6, since this interleukin stimulates C-reactive protein synthesis.
A combination of assays for the labeling index and β2-microglobulin levels is believed to give the best indication of the degree of aggressiveness of a multiple myeloma.
Chemotherapy is the standard treatment for multiple myeloma. Classically treatment consisted of the administration of a combination of melphalan and prednisone in intermit-tent high dosage cycles. Recently, combination chemotherapy regimens (including these two drugs plus several others, such as vincristine, nitrosourea, cyclophosphamide, and dox-orubocin) have gained favor with more complete and durable responses. Even higher rates of remission can be obtained with administration of several chemotherapeutic agents fol-lowed by autologous bone marrow or stem cell transplantation. The preferred approach is to obtain stem cells (CD34+ ) from the peripheral blood after an initial administration of large doses of cytotoxic drugs (particularly cyclophosphamide) plus GM-CSF and use those cells to reconstitute the immune system after more extensive ablation of the bone marrow.
Biological response modifiers have gained popularity in recent years. Administration of interferon-α as maintenance therapy after a favorable response is obtained with chemotherapy has been reported to extend the duration of relatively symptom-free periods. In vitro experiments suggest that interferon-γ inhibits the IL-6–induced growth of some myeloma cell lines and that retinoic acid (an agent known to induce the redifferentiation of malignant cells) depresses the expression of IL-6 receptors on other myeloma cell lines. However, the therapeutic value of these two compounds is still questionable.
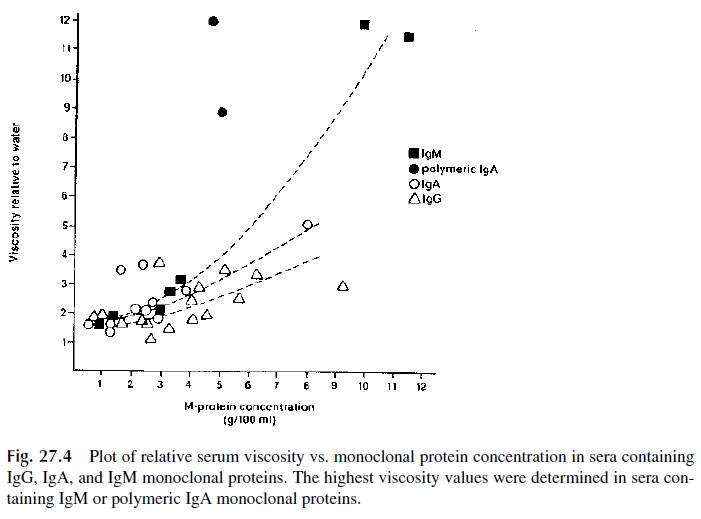
Plasmapheresis, which consists of replacing the patient’s plasma by normal plasma or a plasma-replacing solution, is indicated in cases with hyperviscosity. The reduction of viscosity secondary to the reduction in the circulating levels of monoclonal protein caused by chemotherapy takes time, and the rapid correction of hyperviscosity may be essential for proper management. The rapidly beneficial effects of plasmapheresis are illustrated in Fig. 27.4, which shows the normalization of retinal changes observed after plasmapheresis.
Other supportive measures include hemodialysis or peritoneal dialysis in cases with renal insufficiency, and antibiotic therapy and prophylactic administration of gammaglob-ulin in patients with recurrent infections.
2. Plasma Cell Leukemia
Plasma cell leukemia is the designation applied to cases in which large numbers of plasma cells can be detected in the peripheral blood (exceeding 5–10% of the total white blood cell count). Besides the leukemic picture in the peripheral blood, the remaining clinical and laboratory features of plasma cell leukemia are usually indistinguishable from those of multiple myeloma. The prognosis is generally poor; this may reflect a higher degree of dedifferentiation on the part of malignant plasma cells that abandon their normal territory.
3. “Benign” or “Idiopathic” Monoclonal Gammopathies
The designation of benign or idiopathic monoclonal gammopathy (monoclonal gammopa-thy of undetermined significance, MGUS) is used when a monoclonal protein is found in an asymptomatic individual or in a patient with a disease totally unrelated to B-lymphocyte or plasma cell proliferation (solid tumors, chronic hepatobiliary disease, different forms of non–B-cell leukemia, rheumatoid arthritis, etc.).
Scandinavian authors conducting extensive population studies have given an average figure for the incidence of idiopathic monoclonal gammopathies of about 1%, by far the most common form of B-cell dyscrasia. The incidence seems to increase in the elderly, up to about 20% in 90-year-old and older individuals.
The clinical significance of the finding of a benign or idiopathic monoclonal gam-mopathy lies in the need to make a differential diagnosis with a malignant B-cell dyscrasia in its early stages. Several criteria for the differential diagnosis between benign and malig-nant plasma cell dyscrasias have been proposed (Table 27.2).
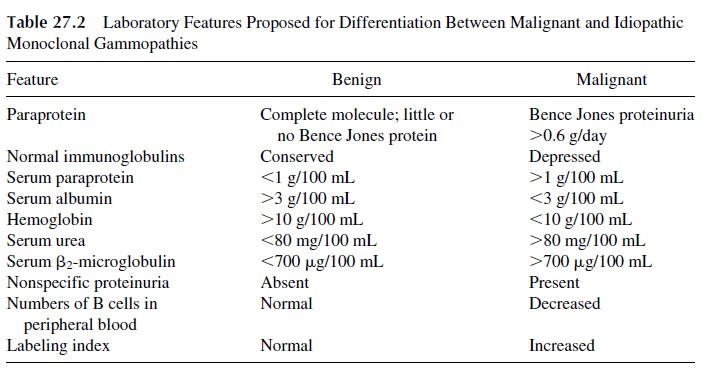
A good practical rule is to assume that any monoclonal gammopathy detected unex-pectedly during the investigation of a condition not clearly related to a B-cell malignancy, or during screening of a normal population (particularly in individuals of advanced age), should be considered as benign until proven otherwise. The best attitude in such cases is to withhold cytotoxic therapy and follow the patients closely, every 3–6 months, measuring the amount of paraprotein; malignant cases show a progressive increase, whereas in benign cases the levels remain stable. Patients with benign gammopathy have to be observed at least yearly after the first 2 years of follow-up, since there are documented cases of malig-nant evolution after 5 or more years of “benign” behavior.
4. Waldenström’s Macroglobulinemia
Waldenström’s macroglobulinemia, a malignancy of lympho-plasmacytoid cells associ-ated with increased levels of macroglobulins, was first described by a Swedish physician, Dr. Jan Waldenström. The macroglobulins were later defined as IgM monoclonal proteins. Thus, the diagnosis of Waldenström’s macroglobulinemia requires proof of the existence of a monoclonal IgM protein in the patient’s serum.
Clinical features. Waldenström’s macroglobulinemia is clinically characterizedby a constellation of symptoms that include weakness and anemia, hyperviscosity-related symptoms (Table 27.1), diffuse osteoporosis, hepatomegaly, splenomegaly, and lym-phadenopathy.
Symptoms suggestive of multiple myeloma, such as bone pain or “spontaneous” frac-tures, are rare. The immunosuppression is also milder than in multiple myeloma. Hyper-calcemia, leukopenia, thrombocytopenia, and azotemia are rarely seen. Renal insuffi-ciency, when present, is usually a manifestation of serum hyperviscosity and can be reversed by plasmapheresis and/or peritoneal dialysis.
Diagnosis.The two main diagnostic features of Waldenström’s macroglobuline-mia are the presence of an IgM monoclonal protein and the pleomorphic infiltration of the bone marrow with plasma cells, lymphocytes, and lympho-plasmacytic cells.
Management.Waldenström’s macroglobulinemia is a disease of old age and fre-quently follows a benign course. The most common life-threatening complications result from serum hyperviscosity. In such cases, repeated plasmapheresis is often sufficient to keep the patient asymptomatic, avoiding the use of cytotoxic drugs and their side effects. Cytotoxic therapy may be required, due to the severity of the symptoms or the impossibil-ity of keeping the patient on repeated plasmapheresis. Chlorambucil (leukeran) is the drug of choice, usually given in a continuous low dosage.
5. The Heavy-Chain Diseases
Some B-cell dyscrasias are associated with the exclusive production of heavy chains (or fragments thereof) or with the synthesis of abnormal heavy chains that are not assembled as complete immunoglobulin molecules and are excreted as free heavy chains. Both types of abnormality can be on the basis of a heavy-chain disease. The heavy-chain diseases are classified according to the isotype of the abnormal heavy chain as γ, α, µ, and δ (one sin-gle case of δ chain disease has been reported, and ε chain disease has yet to be described).
α-Chain Disease. This is the most common and best defined heavy-chain disease.It affects patients in all age groups, even children, and is more frequent in the Mediter-ranean countries, particularly affecting individuals of Jewish or Arab ancestry.
Clinically it is indistinguishable from the so-called Mediterranean-type abdominal lymphoma, characterized by diarrhea and malabsorption unresponsive to gluten with-drawal, with progressive wasting and death. Intestinal x-ray changes are suggestive of dif-fuse infiltration of the small intestine such as thickened mucosal folds. Intestinal biopsy re-veals diffuse infiltration of the submucosa by reticulo-lymphocytic cells.
Diagnosis relies on the demonstration of free alpha chains, usually in serum. Routine electrophoresis usually fails to show a monoclonal component, but immunoelectrophoresis shows an abnormal IgA arc that does not react with antisera specific for light chains
γ-Chain Disease. This was the first form of heavy-chain disease discovered. Clin-ically, it appears as a lymphoma with lymphadenopathy, splenomegaly, and hepatomegaly. Bone marrow and lymph node biopsies show lympho-plasmocytic proliferation. The diag-nosis is based on the immunochemical demonstration of free γ chains in the serum and/or urine.
µ-Chain Disease. This variant of heavy-chain disease is less frequent than either γ or α heavy-chain diseases, and clinically it is indistinguishable from chronic lymphocytic leukemia or lymphocytic lymphoma, with marked Bence Jones proteinuria and small amounts of free µ chains detectable in the serum and sometimes also in the urine.
Case 27.1 Revisited
The history of progressive weakness, malaise, fatigue, with shortness of breath after climb-ing one flight of stairs in 55-year-old woman is suggestive of anemia, supported by the ob-servation of pale conjunctival mucosae. The anemia of multiple myeloma has an obscure etiology and is neither hemolytic nor secondary to iron deficiency.
Progressive loss of vision with bilateral retinal changes (irregularly dilated, tortu-ous veins, with flame-shaped retinal hemorrhages and “cotton wool” exudates) is very sug-gestive of serum hyperviscosity, which is also associated with increased bleeding tenden-cies. Serum hyperviscosity causes venous stasis with exudation and bleeding in the retinal capillaries, which leads to progressive vision impairment.
In a patient with serum hyperviscosity, the main differential diagnosis is between multiple myeloma and Waldenström’s macroglobulinemia, since this last disease is fre-quently associated with the hyperviscosity syndrome. The finding of osteolytic lesions, a very high level of IgA and low IgM strongly indicate multiple myeloma as the most likely diagnosis. Additional laboratory tests in this patient revealed a monoclonal protein of gamma mobility, characterized as predominantly polymeric IgA, normal urine proteins, a serum viscosity of 13.2 relative to distilled water (normal<2), and extensive plasma cell infiltrates in the bone marrow. All these findings were compatible with the diagnosis of multiple myeloma and hyperviscosity syndrome.
The patient was treated with plasmapheresis (two to three sessions a week in each of which 300–500 mL of plasma were exchanged) and cyclophosphamide. A marked clinical improvement of all symptoms related to serum hyperviscosity was seen soon after plasma-pheresis was initiated, and repeated measurements of serum viscosity showed normaliza-tion after one month of combined therapy. After 1 1/2 months of combined therapy, plasma-pheresis was stopped and the patient continued to be treated with cyclophosphamide. Fundoscopy revealed normalization of the retina after 5 months of treatment.
Related Topics