Chapter: Modern Pharmacology with Clinical Applications: Adrenoceptor Antagonists
alpha-Receptor Blocking Agents
α-RECEPTOR
BLOCKING AGENTS
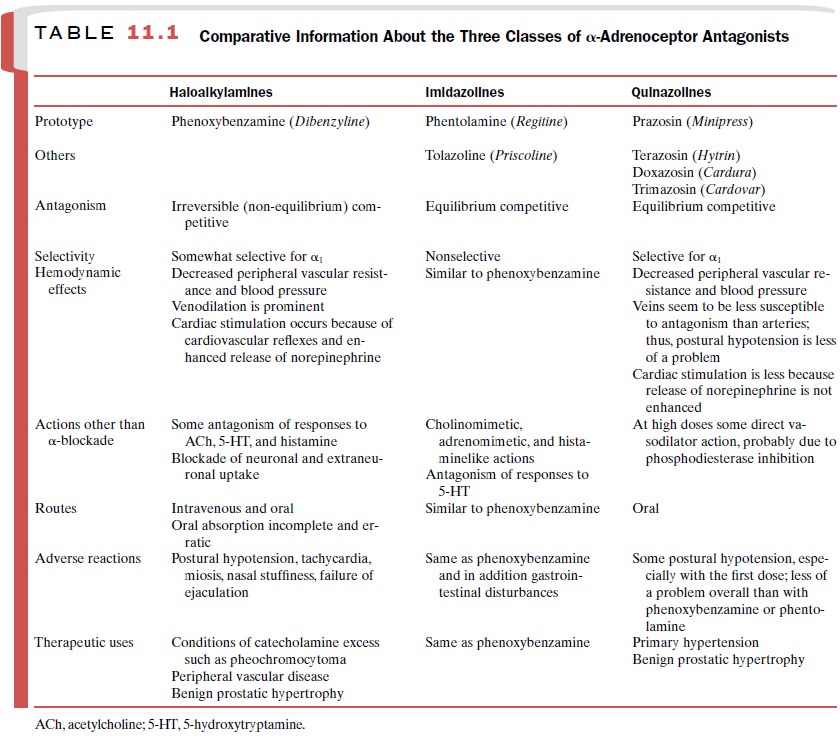
The clinically important α-blockers fall primarily into
three chemical groups: the haloalkylamines
( e.g., phe- noxybenzamine), the imidazolines
(e.g., phentolamine), and the quinazoline
derivatives (e.g., prazosin). Of these three classes of α-adrenoceptor antagonists,
the quina-zoline compounds are of greatest clinical utility. The use of the
haloalky-lamines and imidazolines has diminished in recent years because they
lack selectivity for α1- and α 2-receptors. Comparative information concerning the three chemi-cal
classes of antagonists is presented in Table 11.1
Quinazoline Derivatives
The chief use of these drugs is in the management of pri-mary
hypertension. Examples of quinazoline α-blockers
include prazosin (Minipress),
trimazosin (Cardovar), terazosin (Hytrin), and doxazosin (Cardura).
Mechanism of Action
The -antagonism produced by
prazosin and the other quinazoline derivatives is of the equilibrium-competi-tive type. The drugs are selective for α1-adrenoceptors, so that at usual therapeutic
concentrations there is little or negligible antagonism of α2-adrenoceptors. However,
selectivity is only relative and can be lost with high drug concentrations.
While most of the pharmacological ef-fects of prazosin are directly
attributable to 1-antago-nism, at high doses the drug can cause
vasodilation by a direct effect on smooth muscle independent of -recep-tors.
This action appears to be related to an inhibition of phosphodiesterases that
results in an enhancement of intracellular levels of cyclic nucleotides.
Absorption, Metabolism, Excretion
Prazosin is readily absorbed
after oral administration, peak serum levels occur approximately 2 hours after
a single oral dose, and the antihypertensive effect of pra-zosin persists for
up to 10 hours. Its half-life in plasma ranges from 2.5 to 4 hours, and
elimination from plasma appears to follow first-order kinetics. The drug is
exten-sively (perhaps as high as 97%) bound to plasma pro-teins; this observation
partially explains the lack of cor-relation between plasma drug levels and
persistence of antihypertensive effect.
Hepatic O-dealkylation and glucuronide formation appear to be major
pathways of biotransformation. Only about 10% of orally administered prazosin
is ex-creted in the urine. Plasma levels of prazosin are in-creased in patients
with renal failure; the nature of this interaction is unknown.
Pharmacological Actions
The most important pharmacological effect of prazosin is its ability to antagonize vascular smooth muscle contrac-tion that is caused by either sympathetic nervous activity or the action of adrenomimetics.
Hemodynamically, the effects of prazosin differ from those of phenoxybenza-mine and
phentolamine in that venous smooth muscle is not as much affected by prazosin.
Postural hypotension during chronic treatment is also less of a problem. Also,
increases in heart rate, contractile force, and plasma renin activity, which
normally occur after the use of va-sodilators and α-blockers, are much less
prominent fol-lowing chronic treatment with prazosin.
Phenoxybenzamine and
phentolamine, in addition to blocking postsynaptic α -receptors, also block α 2-receptors on nerves and
therefore can enhance the re-lease of norepinephrine. When norepinephrine
exerts a postsynaptic action by means of β-adrenoceptors (e.g., cardiac stimulation,
renin release), blockade of presy-naptic α2-receptors by phenoxybenzamine and
phentol-amine may actually potentiate the responses. Prazosin blocks responses
mediated by postsynaptic α 1-receptors but has no effect on the presynaptic α 2-receptors. Thus, stimulation of the heart and renin
release is less promi-nent with this drug.
Clinical Uses
Prazosin is effective in
reducing all grades of hyper-tension. The drug can be administered alone in
mild and (in some instances) moderate hypertension. When the hypertension is
moderate or severe, prazosin gen-erally is given in combination with a thiazide
diuretic and a β-blocker. The antihypertensive actions of pra-zosin are
considerably potentiated by coadministra-tion of thiazides or other types of
antihypertensive drugs.
Prazosin may be particularly
useful when patients cannot tolerate other classes of antihypertensive drugs or
when blood pressure is not well controlled by other drugs. Since prazosin does
not significantly influence blood uric acid or glucose levels, it can be used
in hy- pertensive patients whose condition is complicated by diabetes mellitus
or gout.
Prazosin and other
-antagonists find use in the management of benign prostatic obstruction,
especially in patients who are not candidates for surgery. Blockade of α-adrenoceptors in the base of
the bladder and in the prostate apparently reduces the symptoms of obstruc-tion
and the urinary urgency that occurs at night.
Adverse Effects
Although less of a problem
than with phenoxybenza-mine or phentolamine, symptoms of postural hypoten-sion,
such as dizziness and light-headedness, are the most commonly reported side
effects associated with prazosin therapy. These effects occur most frequently
during initial treatment and when the dosage is sharply increased. Postural
hypotension seems to be more pro-nounced during NA+ deficiency, as
may occur in patients on a low-salt diet or being treated with diuretics, β- blockers, or both.
Related Topics