Chapter: Medical Immunology: The Complement System
The Classical Complement Pathway - Medical Immunology
THE CLASSICAL COMPLEMENT PATHWAY
Activation of the Classical Complement Pathway By Antigen-Antibody Complexes
Immunoglobulins and native complement components are normally found in the serum and in the lymph, but these molecules do not interact with each other until the antibodies inter- act with their corresponding antigens and undergo the necessary conformational changes. These immunoglobulin conformational changes are the basis for specific activation of the very powerful classical complement pathway.
1. The Initiation of the Cascade: C1 Activation
Native, free immunoglobulin does not activate the complement system. A single native IgG molecule will not bind and activate the first component (C1) in the complement pathway. However, if IgG antibodies are aggregated by antigen binding, wherein their Fab arms move about the hinge region in order to bind to antigenic determinants and therein expose the CH2γ region on their Fc, this will result in C1 binding (fixation) and activation.
The IgG subclasses vary with regard to their efficiency in activating C1, which is di-rectly related to the length of the IgG hinge region. A longer hinge region allows movement of the Fab arms further away from the Fc so as to more fully expose the CH2 region. Thus IgG3, upon binding antigen, is by far the most efficient subclass of IgG in activating C1, followed by IgG1 and weakly by IgG2. IgG4 does not effectively activate the classical complement pathway.
For C1 to be activated, it must bind to at least two adjacent IgG antibodies that are bound to antigens. This usually means that the concentration of antibody must be relatively high and that the specific antigenic determinants recognized by the IgG antibody must be in close proximity. When antigens are in close proximity on a bacterial or viral surface, C1 is very effectively activated, especially when specific IgG3 or IgG1 antibodies are de-posited. For IgM, a pentamer, these logistical problems are less critical. Indeed, IgM-im-mune complexes are very potent activators of C1. IgA antibodies, in contrast, have rela-tively weak classical pathway activating properties.
2. The Early Stages: C1 to C3
The aggregated Fc regions of the deposited IgG molecules have binding sites, located on the CH2 domain, for an umbrella-like subcomponent of the C1 molecule, known as C1q. Detailed chemical studies have revealed that C1 is actually a complex of three different types of molecules (one C1q molecule, two C1r molecules, and two C1s molecules) held loosely together through non covalent bonds and requiring a physiological Ca2+ concen-tration for their proper association.
Under physiological conditions, the subcomponents of C1 (C1q, C1r2, and C1s2) as-sociate but exist in conformations that partially limit the “tightness” of that association. Physiological levels of calcium ions and ionic strength are essential to maintain these slightly weaker associations within the C1 macromolecular complex. These conditions pre-vent spontaneous C1 activation that would otherwise readily occur upon a tighter association.
C1q contains several distinct portions; a collagen-like stem region, which branches into a six-branched umbrella-shape. Within the umbrella portion of the stem an association with the two C1r and two C1s molecules occur. Each of the six collagen-like branches ter-minates in a single globular head region. It is these globular head regions that have the po-tential to associate with the exposed Fc regions of the antibodies present on immune com-plexes (Fig. 9.1). As a consequence of the C1q globular head regions interacting with the exposed CH 2 of adjacently deposited IgG3 antibodies, C1q undergoes significant confor-mational changes. This dramatic C1q conformational change spontaneously facilitates a tightened C1q-C1r2-C1s2 association, resulting in the movement and self-activation of the
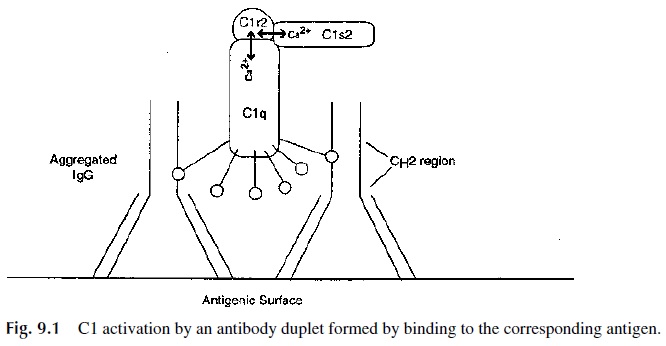
The activated C1r2 enzymes have a protease activity that cleaves a peptide bond within the two adjacent C1s molecules, which in turn become activated en-zymes. Activated C1s2 enzymes (within the C1 macromolecular complex) are then able to cleave and activate the next component in the series, C4.
It is important to realize that at this step active host C1s enzymes are present on the antigenic surface via the association of the immune complexes with activated C1. The de-position of complement enzymes on the antigenic surface is the major reason for the rapid, amplifying effect of the classical complement pathway, because C1s enzymes will continue to activate C4 and C2 until the C1s enzymes are inhibited.
As native C4 molecules come into contact with the C1-immune complex, they bind and are cleaved by activated C1s into a small fragment, which remains soluble (C4a), and a larger fragment, C4b. As a helpful rule remember that fragments released into the fluid phase are often designated by the letter a, while those fragments that remain bound to mem-branes are designated by the letter b. However, the nomenclature of C2 fragments is re-versed, i.e., the bound fragment is designated as C2a, and the soluble fragment as C2b. Each activated C1 is able to cleave and convert many C4 molecules to C4a and C4b. The second fragment derived from C4, C4b, has a very short-lived and highly reactive binding site. This active binding site (an acylating group) allows C4b to bind covalently to the near-est hydroxy or amino group, which is usually located on the antigenic surface (Fig. 9.2). Antibody-coated viral envelopes or antibody-coated bacterial membranes serve as excel-lent sites for C4b deposition.
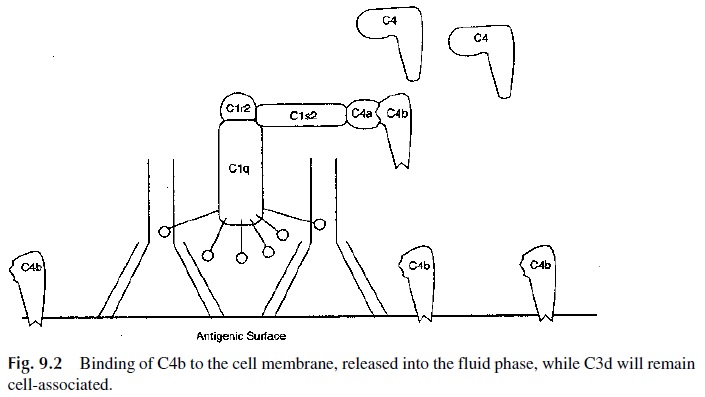
Any activated C4b molecules that do not reach the nearby antigenic surface within a few nanoseconds (unable to bind covalently to the antigen) will lose their short-lived active binding site and undergo conformational changes that facilitate binding to a serum factor termed C4-binding protein. Binding of C4 to C4-binding protein causes rapid loss of C4b function and is a very important control mechanism to protect the host’s tissues from “by-stander attack” by the C4b molecules being formed in areas of infection.
The activated C1s within the bound C1 macromolecular complex is also responsible for the activation of C2, the next complement component to be activated in the classical pathway. In the presence of magnesium ions (Mg2+), C2 interacts with antigen-bound C4b and is, in turn, split by C1s into two fragments, termed C2b and C2a. C2b fragments are re
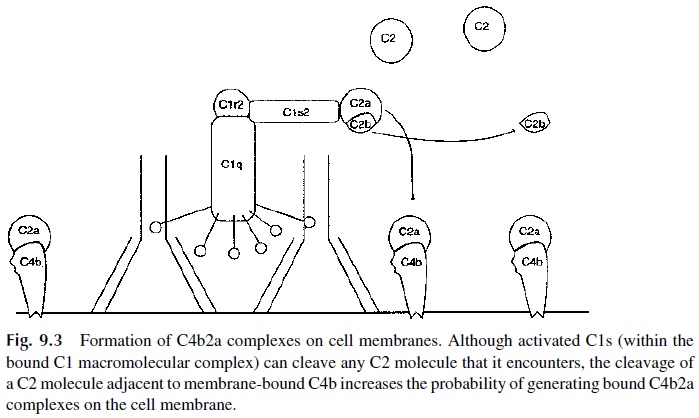
Thus, proper concentrations of Ca2+ ions are needed for optimal C1q-C1r2-C1s2 interactions, and Mg2+ ions are required for proper C4b-C2a formation. In the absence of Ca2+ and/or Mg2+ (due to the addition of metal chelators such as EDTA), the classical activation pathway is interrupted. An exces-sive level of Ca2+ also tends to disrupt the association of C1q-C1r2-C1s2.
The biological role of the soluble C2b fragments is controversial. Some scientists postulate that the soluble C2b fragments induce increased capillary permeability, causing leakage of fluid into the interstitial spaces (edema). Thus, the generation of large amounts of C2b is speculated to be one of several pathogenic events in hereditary angioneurotic edema .
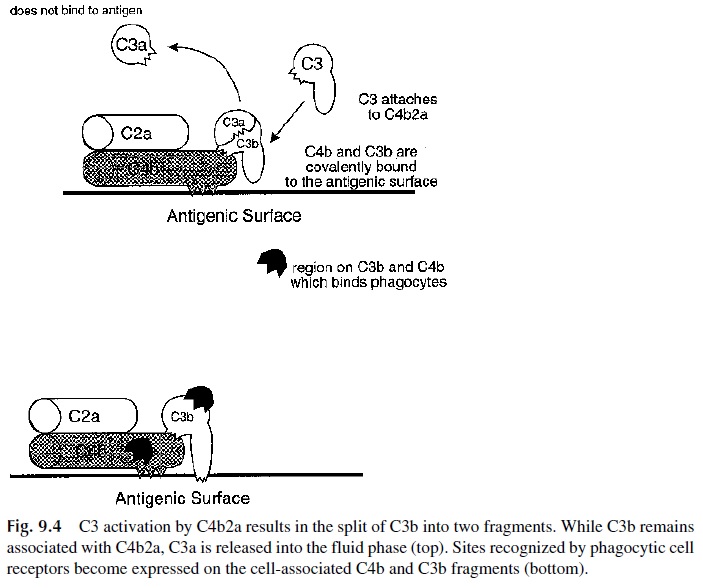
However, the C2 fragment with the best-defined role is C2a, which, when bound to C4b, has the active enzyme site necessary for the activation of C3. For C3 to be activated, C2a must remain as a stable complex associated with the membrane-bound C4b molecule. The active C4b2a complex is also known as C3 convertase because it enzymatically con-verts the next component in the series, C3 to C3b and C3a (Fig. 9.4). Once active C4b2a complexes are deposited on the antigenic membrane surface surrounding each of the im-mune complexes, each bound C4b2a complex is capable of rapidly activating many C3 molecules, until the C4b2a complex is disrupted or the enzymatic activity of C2a decays. When deposited C3b associates with the bound C4b2a to form C4b2a3b the complex ac-quires the capability of binding to C5, wherein C2a (within the C4b2a3b complex) cleaves C5 into C5b and C5a. The newly formed C3b must bind to an amino group or hydroxyl group on the antigen (via its very short-lived active binding site) to remain active. If this fails to happen, the fluid phase C3b associates with Factor H in the serum and is quickly di-gested by a serum protease, Factor I.
Thus, a general rule emerges. As each additional component is added to the antigen-antibody-complement complex, the growing complexes acquire the information needed for binding and activating the next component in the series. The activities expressed at each stage of the sequence are regulated by several mechanisms, including the spontaneous de-cay of C2a activity with time, the short-lived active binding sites on activated complement fragments and the effects of the normally occurring serum complement inhibitors. In general, the roles of the serum complement inhibitors are to:
1. Restrict the complement cascade to the surface of the foreign material
2. Prevent bystander damage to the host
3. Limit unnecessary consumption of complement components
3. Regulatory Mechanisms of the Early Stages
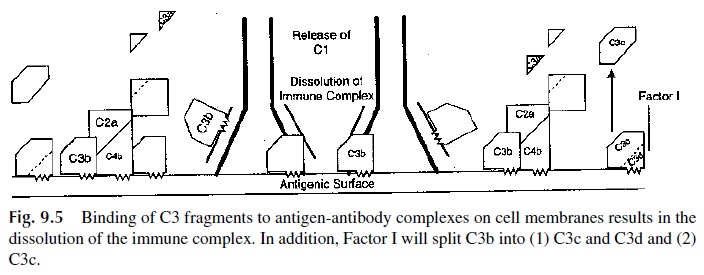
Soon after antigen and antibody react, C4b and especially C3b accumulate on the antigenic surface at such high levels that they begin to deposit onto the specific antigenic determi-nants recognized by the antibody molecules. C4b and C3b molecules also bind to the Fab region of the bound antibodies. Both of these phenomena interfere with the ability of anti-bodies to remain associated with specific epitopes in the antigen. This partial dissolution of the immune complex results in the loosening of the C1 macromolecule from the immune complex as the antibody molecule recovers its native configuration and the complement binding sites on the CH2 region become less accessible. As C1 begins to separate from the immune complex, the C1q-C1r2-C1s2 macromolecular complex tends to return to its loosely associated form. At that point, the activated C1r2 and C1s2 enzymes are extremely susceptible to irreversible inhibition by a normal serum glycoprotein termed C1 inhibitor (C1 INH). C1 INH forms C1-INH-C1r and C1-INH-C1s complexes, most of which are sep-arated from the bound C1q (Fig. 9.5). Activated C1, once having performed its function while bound to the immune complex, is now irreversibly inhibited from unnecessarily con-suming more native C4 and C2. The proper function of C1-INH represents a very impor-tant regulatory mechanism that restricts the range of activated C1 action to the surface of the antigen and prevents useless consumption of C4 and C2.
4. Immune Adherence and Phagocytosis
Prior to its decay or its inactivation, each of the activated membrane-bound activated C4b2a enzyme complexes activates many C3 molecules. The normal concentration of serum C3 is about 130 mg/dL, which is relatively high and indicates the importance of C3 in the complement activation pathways. C3 is converted through a process that involves its proteolytic cleavage and release of a small biologically active peptide, termed C3a. The larger C3b fragment upon activation behaves very much like the C4b fragment, in that it also has a short-lived, highly reactive binding site, which binds irreversibly to the nearest membrane surface, which is usually the complement-activating antigen.
C3b associates with the bound C4b2a complexes. However, many other C3b molecules bind independently to the antigenic membrane. The deposited C3b molecules also participate in a tremendous amplification of additional C3b deposition via a mecha-nism termed the amplification loop. It is important to understand that the irreversible (covalent) binding of C4b and of C3b molecules to the anti-gen actually changes the nature of the antigen. In the case of endotoxin, C4b and C3b de-position abrogates the toxicity of the molecule. Similarly, C4b and C3b contribute to anti-body-mediated neutralization of viruses, rendered incapable of properly binding and infecting host cells.
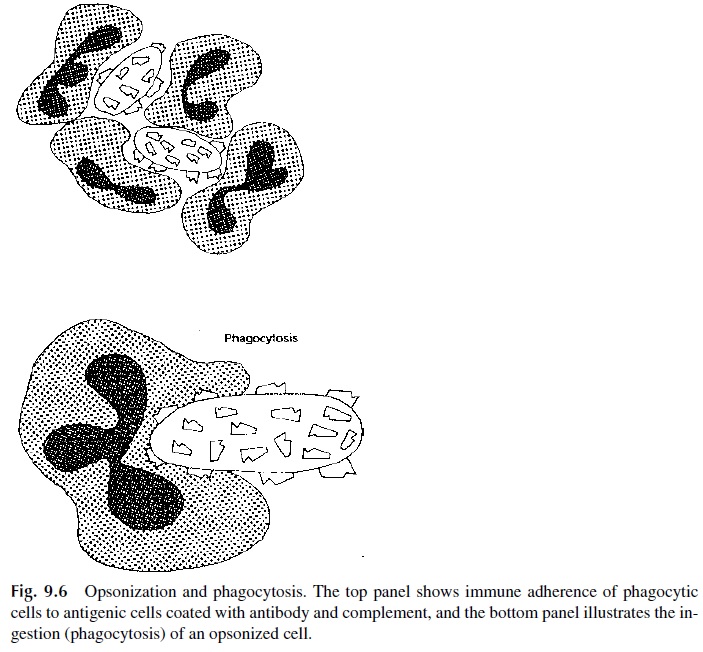
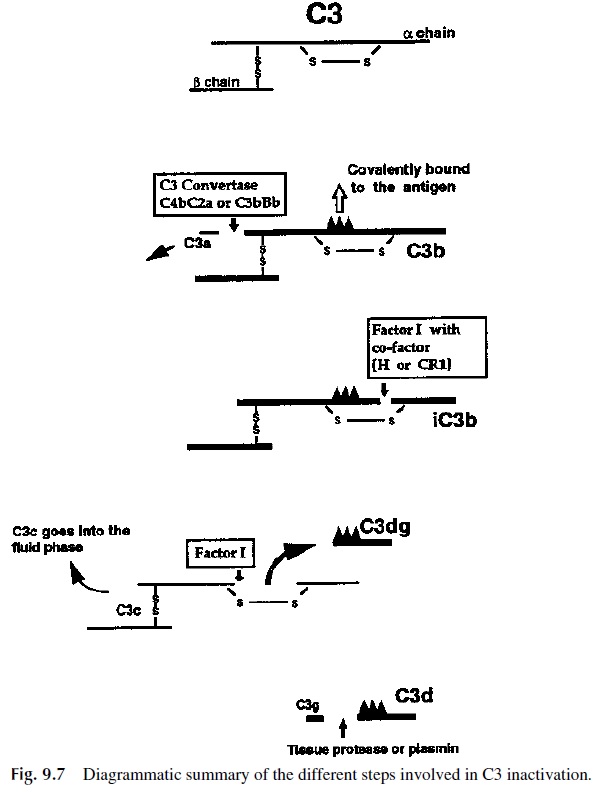
Of great biological significance is the fact that after C4b and C3b bind to the antigenic surface, they undergo conformational changes that result in the exposure of regions of these two molecules that extend away from the antigenic membrane. These exposed parts of C4b and C3b are biologically important because they contain molecular segments which are able to bind to C3b/C4b receptors (currently designated as CR1, complement receptor 1) located on host phagocytic cells (Fig. 9.6). Additional types of receptors for other regions on bound C3b play also a significant role in enhancing phagocytosis. Some of those receptors recog-nize interior (cryptic) regions of C3b exposed as this component is further catabolized (with time) to form inactivated C3b (iC3b), then C3dg, and finally C3d (Fig. 9.7).
Polymorphonuclear leukocytes, like other phagocytic host cells, have thousands of complement receptors on their membranes allowing them to bind, with high avidity, parti-cles coated with C3b (and C4b) and/or with breakdown products of bound C3b (i.e., iC3b). This increased avidity of phagocytic cells for complement-coated particles is known as en-hanced immune adherence, and its main consequence is a significant potentiation of the phagocytosis process. In this process, the phagocyte is stimulated to engulf the comple-ment-coated particle because of the interactions with complement receptors on the phago-cytic cell membrane. Once engulfed, antigens are digested in phagolysosomes, vesicles that result from the fusion of phagosomes, containing phagocytosed particles, and lysosomes, which contain a large variety of degradative enzymes. The phagocytic process is one of the most important fundamental defense mechanisms because it provides a direct way for the host to digest foreign substances .
5. The Late Stages: C5 to C9
The full effect of the activation of the later complement components is evident when the activated complement components are deposited on a cell membrane. C5 molecules can be activated by antigen-bound (membrane-associated) C4b2a3bn complexes or by alternative pathway/amplification loop enzymes, to be discussed later. As expected, activation of C5 is mediated by the specific proteolytic cleavage of C5 molecules by activated C4b2a3bn de-posited on the antigen (e.g., foreign cell membrane). Each C5 molecule first binds to an ac-tivated C4b2a3bn complex and then is split into a small fragment (C5a), which is released into the fluid phase, and a large fragment (C5b). Unlike other complement fragments pre-viously discussed, C5b does not bind immediately to the nearest cell membrane. A com-plex of C5b, C6, and C7 is first formed, and then the C5b67 complex attaches to the cell membrane through hydrophobic amino acid groups of C7, which become exposed as a con-sequence of the binding of C7 to the C5b-C6 complex (Figs. 9.8 and 9.9). The membrane-bound C5b-6-7 complex acts as a receptor for C8 and then C9. C8, on binding to the com-plex, will stabilize the attachment of the complex to the foreign cell membrane through the transmembrane insertion of its alpha and beta chains and attracts C9.
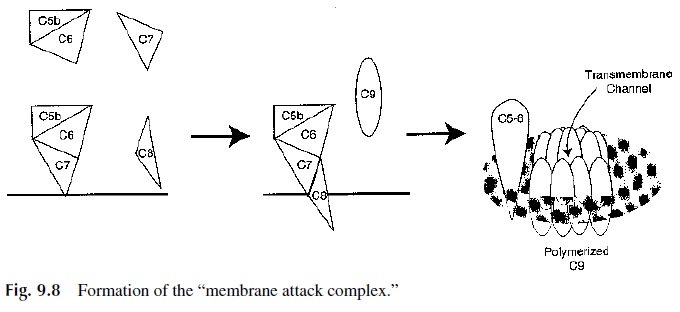
The entire C5b-9 complex is also known as the membrane attack complex (MAC). This designation is due to the fact that on binding to C5b-8, C9 molecules undergo polymeriza-tion, forming a transmembrane channel of 100 Å diameter, whose external wall is believed to be hydrophobic, while the interior wall is believed to be hydrophilic. This transmembrane channel will allow the free exchange of ions between the cell and the surrounding medium. Due to the rapid influx of ions into the cell and their association with cytoplasmic proteins, the osmotic pressure rapidly increases inside the cell. This results in an influx of water, swelling of the cell, and, for certain cell types, rupture of the cell membrane and lysis.

Less than 20 seconds is required for lysis of 1 million sheep erythrocytes coated with excess IgG antibody when they are mixed with 1 mL of fresh undiluted human serum as a source of complement. In contrast, many gram-positive bacteria are not susceptible to dam-age by the MAC as long as their membrane is covered by an intact cell wall. For these or-ganisms, complement-mediated, enhanced phagocytosis is of prime importance.
Normal human cells are somewhat resistant to lysis by human complement. Human cells express substances on their membranes that effectively inhibit the human complement sequence (but not the complement of other species). In addition, phagocytes quickly endo-cytose and destroy inadvertently deposited membrane-bound complement components.
One of the important inhibitory substances on most types of host cells is the CR1 re-ceptor glycoprotein, which in binding to activated C3b, blocks C3b function in the com-plement sequence by causing C3b to be rapidly cleaved to inactivated C3b (iC3b), by a serum enzyme known as Factor I. Obviously, once the phagocyte is actively engulfing the complement-coated particle there is no reason to continue consuming additional comple-ment or risk inadvertently damaging the phagocyte by depositing C3b and/or MAC com-plexes on its surface. Therefore cell surface CR1 molecules after binding to C3b have two independent functions:
· Enhanced phagocytosis of C3b/C4b coated particles by phagocytes
· Inactivation of any C3b/C4b that may become inadvertently deposited on host cells
Decay-accelerating factor (DAF, CD55) is another important complement-inhibitory substance located on a large variety of host cell membranes. The name of this factor derives from the fact that it can accelerate the dissociation of active C4b2a complexes, turning off their ability to continue activating native C3. In addition, DAF attaches to membrane-bound C4b and C3b and prevents the subsequent interaction of C4b with C2a and of C3b with Factor B, respectively. The role of Factor B in the alternative pathway/amplification loop will be explained later. As a consequence, the two types of C3 conver-tases, C4b2a and C3bBb, will not be formed or will become dissociated, and the rate of ad-ditional C3 activation will be limited. Thus the host cell will be spared from complement-mediated membrane damage.
Several other important complement inhibitory substances are also located on almost all host cell surfaces. These substances are protectin (CD59, which restricts formation of the membrane attack complex by binding to C8 and C9) and membrane cofactor protein (CD49, which serves as a cofactor for Factor I enzymatic degradation of C3b).
The existence of these multiple protective mechanisms on host cell membranes ex-plains how a phagocyte approaching a complement-activating immune complex is itself re-sistant to bystander damage initiated by complement inadvertently deposited on its surface. On the other hand, overexpression of complement inhibitors on malignant breast cancer cell membranes and malignant endometrial tissues suggests that avoidance of complement-mediated damage allows better survival of these cancers.
6. Important Biologically Active Fragments: C5a and C3a (Anaphylatoxins and Chemotactic Factors).
The small complement fragments, C5a and C3a, released into the fluid phase are recog-nized by neutrophils and cause these phagocytes to migrate in the direction from which these small fragments originated. The term for this chemical attraction is chemotaxis, and its main biological function is to attract phagocytes into a tissue in which a complement-activating antigen-antibody reaction is taking place. Once the PMNs reach the area, by moving towards the highest concentration of chemoattractants, they will bind to the C4b and C3b coated antigenic substance via their CR1 receptors (and to iC3b via their CR3 re-ceptors) and proceed to phagocytize the foreign material.
Besides their role as chemokines, C5a and C3a activate the phagocytic cells that carry C5a and C3a receptors. In the case of neutrophils, such activation leads to the expression of CAMs and facilitates extravascular migration. In the case of circulating basophils and mast cells located in subepithelial and submucosal tissues, C5a and C3a stimulate the re-lease of biologically active mediators such as heparin and vasoactive amines (e.g., his-tamine). The release of histamine into the tissues results in increased capillary permeabil-ity and smooth muscle contraction. Fluid is released into the tissue, causing edema and swelling. There is some evidence that the complement fragments C3a and C5a may also act directly on endothelial cells, causing increased vascular permeability. The end result is very similar to the classical anaphylactic reaction that takes place when IgE antibodies bound to the membranes of mast cells and basophils react with the corresponding antigens . For this reason, C3a and C5a are known as anaphylatoxins.
Related Topics