Chapter: Essential Clinical Immunology: Immunological Aspects of Skin Diseases
Psoriasis - Skin Diseases
PSORIASIS
Psoriasis vulgaris is an inflammatory disease of the skin that affects 2–3 percent of people across North America and Europe. Until recently, this disease was thought to be an abnormality in skin cell differ-entiation, resulting in keratinocyte hyper-proliferation. However, work begun in the late 1970s showed evidence that immune
Selective targeting of activated T cells and immune-related cytokines was then shown to reverse psoriasis in a large num-ber of cases. It is now believed that the epidermal hyperplasia is a consequence of the immune activation of the focal skin lesions. We consider that psoriasis and atopic dermatitis are the most prevalent T-cell-mediated inflammatory diseases in humans.
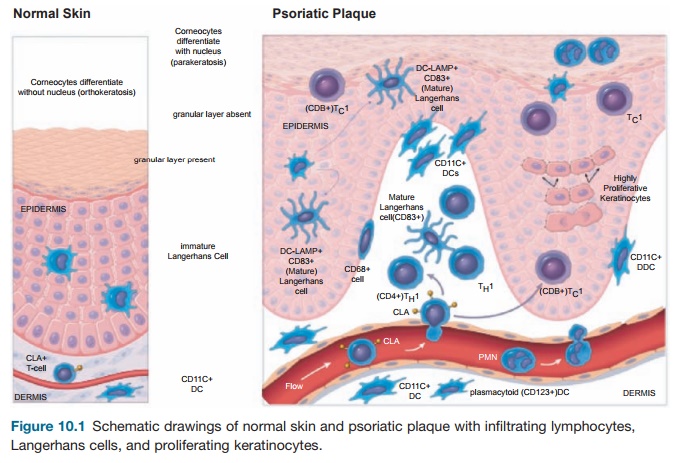
To understand the cellular features of the psoriasis plaque, a clinical and patho-logical description of the lesion is war-ranted. Clinically, the plaque is a red, raised, and scaling (flaking) region of the skin ≥1 cm in size. Usually, many indi-vidual lesions cover the skin separated by normal skin. In extreme cases, virtually all of the skin surface can be affected. Histo-logical features include (1) marked thicken-ing of the epidermis with rete elongation, keratinocyte hyperplasia, and incomplete terminal differentiation of keratinocytes (parakeratosis); (2) infiltration of skin lesions by many types of leukocytes; and (3) increased vascular growth (angiogen-esis) and dilation of blood vessels (Figure 10.1). Leukocyte alterations in psoriatic skin are extensive and include marked numbers of type I T cells (TH1) and DCs, both activated and nonactivated subsets of each cell type, and neutrophils are present in the stratum corneum in most cases. T cells are mainly skin-homing CLA+ mem-ory cells, either CD4+ TH1 or CD8+ TC1 cells. A subset of CD8+CD103+ T cells is further specialized for epithelial homing through surface expression of the E-cad-herin binding integrinαeβ7. Few “allergic” type II T cells (TH2) cells exist in psoriasis skin lesions. Instead, strong TH1 differen-tiation is suggested from genetic profiling
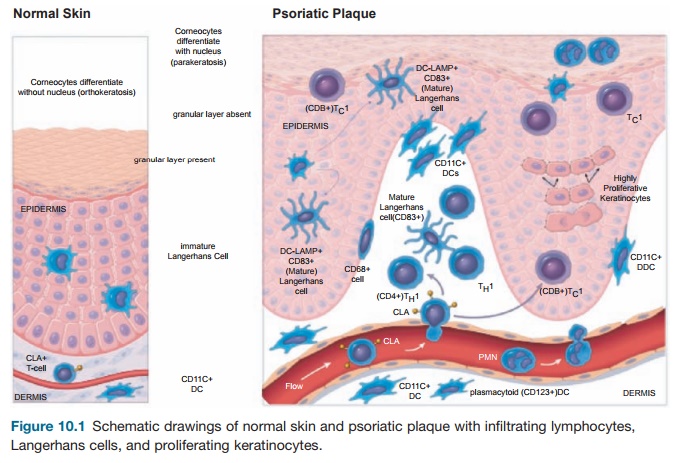
Figure 10.1 Schematic drawings of normal skin and psoriatic plaque with infiltrating lymphocytes, Langerhans cells, and proliferating keratinocytes.
of psoriatic lesional skin T cells compared with normal, including increased expres-sion of GATA-3 and STAT-1 transcription factors and TH1-deviating inflammatory cytokines. Furthermore, systemic immune deviation in psoriasis patients is evidenced by the threefold increase in circulating blood TH1 cells compared with normal controls.
On the basis of finding clonal popula-tions of T cells in psoriatic skin lesions, it has been hypothesized that pathogenic T cells are reactive to as yet an unidentified cutaneous antigen(s) and that the process of generating skin-homing effector T cells is initially the same as a normal cellular response. The formation of psoriatic lesions differs from lymphocytic infiltration in acute hypersensitivity reactions in that activation does not resolve spontaneously (as, for example, after the elimination of an infectious agent). Furthermore, chronic lesions contain a significant infiltration of neutrophils, which is unusual for a “pure” T-cell-mediated response in the skin.
An alternative hypothesis is that, in the genetically programmed psoriatic patient, initial T-cell activation is not necessarily a reaction to a cutaneous antigen but rather to bacterial antigens known to be immu-nologically cross-reactive with keratino-cytes such as the group A streptococcus. Evidence for this pathway is suggested by an elevated immune response to strepto-coccal antigens in guttate psoriasis and the presence of antigens similar to streptococ-cal M proteins in the psoriatic lesions.
The model in Figure 10.1 considers mainly the pathogenic activity of T cells in disease pathogenesis, but one must con-sider the influence of the innate immune response on chronic cellular activation in psoriasis plaques. Yet, the T-cell model has been a working hypothesis on which the therapeutic development of immune-targeted biological drugs has been based.
The use of various animal models, but most especially transplanted human skin on immunodeficient mice, has also been helpful in our understanding of the patho-genic mechanisms involved in the disease.
Animal Models
At the onset, there is no spontaneous model of psoriasis in animals. However, the use of genetically engineered mice or the implan-tation of psoriasis plaque xenografts in immunodeficient mice has aided greatly in understanding some of the basic mecha-nisms of skin inflammation as they apply to psoriasis and other inflammatory diseases.
Several types of transcription factors, including STAT-1 and interferon (IFN) reg-ulatory factors (IRFs), convert IFN-induced genes. IRF-1 is a transcriptional activator, whereas IRF-2 suppresses IRF-1 activity. The IRF-2 null mouse chronically overex-presses IRF-1. These mice spontaneously develop inflammatory skin lesions similar to psoriasis, including CD8+ T-cell infiltra-tion in the epidermis, CD4+ T cells in the der-mis, and marked epidermal hyperplasia.
IL-12 and IL-23 (related cytokines that share a p40 subunit) stimulate T cells to differentiate and produce IFN-γ. Mice were genetically engineered to express constitu-tively p40 in the skin by joining the kera-tin-14 promoter with the p40 gene. These animals developed inflammatory skin lesions with marked epidermal hyperpla-sia and increased levels of DCs and T-cell-derived cytokines similar to those found in psoriasis lesions. Thus, IL-23 appears to be an important “upstream” inflammatory product leading to possible IFN-γ produc-tion and synthesis of “downstream” genes controlled by IFN-γ, STAT-1, and IRFs.
Another approach that has yielded useful information has been to transplant unaffected (nonlesional) skin or lesional skin from a psoriasis patient on severe combined immunodeficiency (SCID) mice. Injection of cytokines into the uninvolved tissue grafts induces some mild hyperpla-sia, whereas injection of superantigen-acti-vated mononuclear leucocytes obtained from peripheral blood of the same patient induces a full psoriasis phenotype (but without neutrophil infiltration), suggest-ing that the psoriasis phenotype can be induced in genetically predisposed skin by bacterial antigen-primed leucocytes. When psoriasis lesional skin is grafted, long-term grafts continue to show viable T cells and other infiltrating mononuclear leucocytes. Thus, it appears that viable T cells can con-tinue to expand in situ in skin lesions at a rate that matches the rate of programmed cell death, and one does not need new T cells from the peripheral circulation to perpetuate the lesion.
Perhaps of greater importance has been the observation that when nonlesional “normal” skin from psoriasis patients is grafted onto highly immunodeficient AGR129 mice, the bystander T cells in the graft expand in situ and cause a full-blown psoriatic lesion. Unlike SCID mice, AGR129 mice lack both natural killer (NK) cells and IFN receptors, which may leave them unable to reject graft T cells or create a cytokine environment more conducive to T-cell activation. Either a tumor necrosis factor (TNF) antagonist or anti-CD3 anti-bodies can block the onset of lesions.
Global View of Psoriasis Through Genomics
Another informative approach to under-standing the underlying mechanisms of pathogenesis of the psoriatic plaque has been the study of differences in gene expression detected through transcrip-tional profiling on 63,000-element gene arrays. Using this technique, it appears that 1,338 genes have altered expression in psoriasis. Having a global view of these differences in gene expression between psoriatic plaques and normal skin back-ground is important because it provides an unbiased means to assess activation pathways in psoriasis. Through genomic analysis, new inflammatory or regulatory cytokine and chemokine products have been identified as overexpressed in psori-atic lesions. Unexpectedly, there is strong expression of numerous lymphoid-orga-nizing chemokines, for example, CCL19, CCL21, and SDF-1 in psoriasis, as expres-sion is normally confined to lymph nodes or formal lymphoid tissues. Most likely, these chemokines orchestrate a striking accumulation of immature dendritic cells (iDCs) and mature dendritic cells (mDCs) in skin lesions; T-cell activation in situ may be regulated through interaction with these DCs in the skin or through release of acti-vating cytokines (such as IL-23) produced by these infiltrating DCs. If one considers that the vascular changes in psoriasis bear a striking resemblance to those in lymph nodes, then the picture of dense perivas-cular DC/T-cell aggregates and increased expression of lymphoid chemokines sug-gests that the dermal infiltrates are actually a type of secondary lymphoid tissue.
Genomic profiling permits us to iden-tify which of the many cytokines detected in psoriasis plaques are transcriptional activators. Thus, expression of more than sixty-five genes with increased expres-sion in psoriasis lesions can be linked to IFN-γ and subsequent activation of STAT-1 signaling by this cytokine. For example, the chemokines CXCL9/MIG, CXCL10/IP-10, and CXCL11/I-Tac are STAT-1-regulated
products and are synthesized in large part by keratinocytes in plaques. These che-mokines direct CXCR3+ T cells to migrate to the epidermis, where they may trigger epidermal hyperplasia as a result of physi-cal damage (disruption of basement mem-brane and desmosomes) done to epider-mal structure through T-cell trafficking or through secreted products.
Interleukin-8 (also induced by IFN-γ) is a key regulatory chemokine for neutrophil trafficking into lesions. Inducible nitric oxide synthase (iNOS) transcription is also regulated by IFN-γ and is highly overex-pressed in psoriasis lesions, so that its prod-uct (NO, or nitric oxide) may be responsible for either cell damage or vascular dilation in lesions. Hence, IFN-γ plays a key role in leukocyte migration, as well as in epider-mal and vascular alterations. The T cells in skin lesions do not become activated to release TNF, IFN-γ, and other cytokines unless triggered by antigen recognition, specific cytokines, or both. Hence, both the clonal nature of these T-cell infiltrates in psoriasis and the ability of CTLA4-Ig to reverse disease activity argue for an ongo-ing activation of T cells via classical TCR engagement and co-stimulation. Two cyto-kines produced by activated DCs, IL-12 and IL-23, have been detected in elevated levels in psoriasis lesions. Both of these factors augment IFN-γ production from T cells and may stimulate excessive expan-sion of TH1-biased clones. Therefore, in this model, the presence of activated DCs in skin lesions may be as important in sus-taining disease activity as are T-cell infil-trates. Long-term disease persistence in the skin (individual plaques can last for years without suppression by therapy) has been hypothesized to be a consequence of orga-nized lymphoid tissue that forms in these inflammatory skin lesions.
Insights into Psoriasis Pathogenesis from Treatments
In 2003, on the basis of many observa-tions that T cells play an important role in the pathogenesis of psoriasis, two T-cell-targeting biologics, alefacept and efali-zumab, were initially tested for activity in psoriasis and are now approved by the U.S. Food and Drug Administration (FDA). Ale-facept is a fusion protein that contains the extracellular domain of LFA-3 fused with immunoglobulin constant region domains. This agent binds to CD2, which is expressed at high levels on memory T cells. Although this binding interaction with CD2 could inhibit normal LFA-3/CD2 signaling, suc-cessful therapeutic outcomes with this agent are most closely associated with depletion of T cells from psoriasis skin lesions and, to a lesser extent, depletion of memory T cells (CD8 > CD4) from the circulation.
Efalizumab is a humanized monoclonal antibody that binds to the alpha subunit (CD11a) of the integrin lymphocyte func-tional antigen (LFA-1). LFA-1 is expressed at high levels on T lymphocytes, while other integrins are specific for monocytes, macro-phages, neutrophils, and DCs. Thus, LFA-1 blockade with efalizumab, specifically targets T cells, and alters T-cell function in several ways: (1) prevents firm adhesion of T cells to inflamed (ICAM+) endothelium and thus blocks entry of cells from the cir-culation; (2) prevents binding of T cells to ICAM+ keratinocytes, thus reducing traf-ficking of cells into the epidermis; and (3) reduces T-cell activation, with attendant cytokine release, either directly or by block-ing DC/T-cell immune synapse formation.
Although alefacept and efalizumab are potent immune therapies, only 25–30 percent of treated patients experience maximal disease improvement after a twelve-week treatment course. For unknown reasons, responses to these tar-geted agents are more variable than to more general immunosuppressive treat-ments. Possible explanations include vari-able expression of redundant T-cell activa-tion pathways, restrictions in the access of large molecules to relevant T-cell pools, or more complex interactions between T cells and other types of leukocytes that contrib-ute to disease pathogenesis in different patients. Genetic/genomic heterogeneity of humans seems likely to underlie the vari-able response to molecule-specific antago-nists, but more consistent improvements in disease are seen with less selective T-cell antagonists, for example, cyclosporine, suggesting that psoriasis is fundamentally caused by cellular immune system dis-regulation. In general, suppression of T-cell-mediated inflammation – functionally defined as genomic suppression of IFN-γ, STAT-1, and downstream genes regulated by STAT-1 in psoriasis skin lesions – leads to objective improvement or clearing of disease, whereas failure to suppress these inflammatory genes leads to continuing disease activity. A review of therapeutic trials with many different agents indicates that there are no documented cases in which T cells and associated inflammatory genes were reduced without correspond-ing improvements in disease-defining epi-dermal hyperplasia. Hence, no data argues against the fundamental hypothesis that activation of the cellular immune system triggers psoriasis.
Another therapeutic approach in pso-riasis has been to antagonize inflamma-tory cytokines. Although an antibody to IL-8 was administered to psoriasis patients with moderate improvement in disease activity, the first major success of the anti-cytokine approach was antagonizing TNF
with the chimeric antibody infliximab. Subsequently, etanercept, a TNF/lympho-toxin antagonist fusion protein, consist-ing of the extracellular domain of TNF-R2 and immunoglobulin constant region domains, was shown to improve psoriasis. This agent is now an FDA-approved drug for treating psoriasis and psoriatic arthri-tis. The success of these trials suggests the need to consider psoriatic inflammation in a broader context than just IFN-γ release from activated T cells. Although TNF is often considered a key cytokine of innate immune responses, activated type I T-cells co-synthesize IFN-γ and TNF. Many dif-ferent inflammatory genes, including IL-8 and iNOS, which are central mediators in psoriasis, have composite promoters for both STAT-1 and NFκB transcription fac-tors that are activated by IFN-γ and TNF, respectively. Thus, the extent to which many type I genes are transcribed may be determined by combined levels of TNF and IFN-γ in the lesions. A recent study shows that etanercept induces strong suppression of type I inflammatory genes in psoriasis lesions, consistent with the view that T-cell-mediated pathways are being modu-lated by TNF inhibition. One should also add that TNF stimulates differentiation and activation of DCs, cells that present antigen to T cells. Thus, interference with DCs may break DC/T-cell immune synapses neces-sary for psoriasis pathogenesis.
Pathogenic Insights Provided by Genetics
Several genetic susceptibility loci have been associated with an increased risk of developing psoriasis, but the identity of susceptibility genes has been elusive. Finally, a susceptibility region (PSORS2) has been mapped to a discrete DNA sequence. PSORS2 is shown to encode a mutated binding site for the transcrip-tion factor RUNX1. Two adjacent genes, SCL9A3R1/EBP50 and RAPTOR, each associated with activation-related signal transduction, may be affected in a way that leads to T-cell activation or keratino-cyte hyperplasia, but more work is needed to understand fully the functional conse-quences of this mutation.
In summary, psoriasis vulgaris can be viewed as a cell-mediated autoimmune dis-ease characterized by interaction between DCs, T cells, and inflammatory cytokines. In this reaction, there is transcriptional activation of a broad set of inflammation-producing gene products that change traf-ficking of leukocytes and growth patterns of resident skin cells. Although evidence suggests that type I T cells are probably reacting with an autoantigen in diseased skin, this is not yet proven. Alternatively, the psoriasis could be a disease of an over-active innate immune system or underac-tive T regulatory pathways. The use of animal models, the study of cell types in the lesions, the use of genomics to detect transcriptional profiling, and clinical trials have all been useful in understanding the disease process; however, a good deal of work still needs to be done to treat this seri-ous disease of the skin.
Related Topics