Chapter: Essential Clinical Immunology: Immunological Aspects of Skin Diseases
Antibody Induced Bullous Skin Lesions
ANTIBODY-INDUCED BULLOUS SKIN LESIONS
This group of skin diseases includes pemphigus vulgaris, bullous pemphigoid, der-matitis herpetiformis, and several other relatively rare blistering disorders. Although uncommon, these are serious, sometimes fatal, skin diseases. The group is unified by production of autoantibodies to different adhesion proteins/structures within the epidermis or basement membrane zone at the epidermal–dermal junction.
Pemphigus Vulgaris
This is the most serious of these disorders, and before the introduction of steroids, it was often fatal. The usual age of onset is between forty and sixty years old, but it may occur in any age group. Clinically, it often begins with ulceration of the oral mucosa followed by formation of wide-spread flaccid weepy bullae. Most patients with Pemphigus vulgaris have circulating antibodies to desmosomal adhesion mole-cules such as desmoglein 3, which disrupts intercellular connections within the epider-mis. This results in the loss of normal epi-thelium and detachment of cells. The titer of this antibody often but not always cor-relates with disease activity.
Direct immunofluorescence of the per-ilesional skin is diagnostic and the IgG class of antibodies and C3 complement are seen at the site of the lesions (Figure 10.3). Further support for the direct role of auto-antibodies in this disease is the fact that sera from pemphigus patients will pro-duce pemphigus-like lesions in monkeys and mice. Furthermore, IgG fractions from these sera will induce epithelial cell detach-ment in human skin cultures.
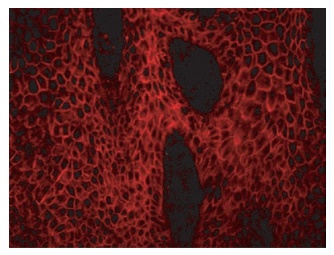
Figure 10.3 Immunofluorescent photograph of a frozen section of psoriasis plaque using serum from a patient with pemphigus vulgaris. Note the clear binding of anti-desmoglein-3 antibodies in the patient’s serum binding to desmoglein-3 protein that is present in desmosomes cementing epidermal cells together.
must be high to suppress new lesions and may reach 100 mg/day. Once new blister formation is under control, the dose can be attenuated. Other forms of antibody sup-pressive treatments have been used, such as azathioprine and low doses of metho-trexate, and even plasmapheresis has been successful in removing circulating anti-bodies, especially if the disease becomes steroid resistant. More recently, deple-tion of B cells with the monoclonal CD20 antibody rituximab has shown efficacy in difficult cases.
Bullous Pemphigoid
The condition is clinically similar to pem-phigus, but the blisters are subepidermal not intraepidermal. This disease occurs more often in an older age group (usually sixty years or older) and is characterized by large tense bullae on thighs, arms, and abdomen. IgG immune complexes and C3 are visualized by indirect immunofluores-cence as a continuous linear band along the basement membrane, detectable in 75–90 percent of patients with active disease.
Although it is believed that autoantibod-ies to a basement membrane protein cause these blisters, experimental demonstration of this association is more difficult than with pemphigus. Sera from these patients do not produce pemphigoid lesions in monkeys and mice. Second, the antibodies remain detectable during remission, and the titers do not correlate with disease activity. Treat-ment is similar to that described for pem-phigus, but the doses needed to suppress lesions are usually lower.
Dermatitis Herpetiformis
The lesions in this condition are usually smaller than those seen in pemphigus, are extremely itchy, and are seen on exte-rior surfaces of elbows, knees, buttocks, necks, and shoulders. Visually, the lesions resemble herpetic papules; however, the herpes virus is not involved in this condi-tion. The age group is quite different from that of pemphigus (twenty-five to forty years old) but it may occur at any age. IgA is the major class of antibody detected, deposited in a granular fashion in the tips of the dermal papillae, forming subepi-thelial bullae. However, unlike pemphi-gus, no circulating autoantibodies to skin are seen in the sera of these patients. An interesting and as yet unexplained obser-vation is that many of the DH patients have a concomitant enteropathy similar to celiac disease. Clinically, they do not have the symptoms of celiac disease and the associated condition is usually only demonstrable by jejunal biopsy. About 30 percent of these patients have antibodies to gliadin and endomysium in their sera. They also have an increased risk of lym-phoma (like celiac disease patients) and a genetic component exhibited by a mark-edly increased inheritance of HLA-B8, DR3-DQ2 haplotypes.
Dermatitis herpetiformis responds to a strict gluten-free diet, which may need to be in force for several years before the lesions heal. Reintroduction of gluten results in the reappearance of the lesions, indicating the important role of gluten in the condition. Yet the presence of IgA deposits in the skin and their immunologi-cal basis remain unknown.
Related Topics