Chapter: Essential Clinical Immunology: Immunological Aspects of Skin Diseases
Alopecia Areata
ALOPECIA AREATA
Alopecia
areata (AA) is most likely a
T-cell-mediated disease of a skin appendage, the hair follicle, and thus can be
classified as an inflammatory skin disease. The condition is quite common
because about 1.7 percent of the population experience an episode of AA during
their lifetime. AA may be focal, affecting only one small region of the skin
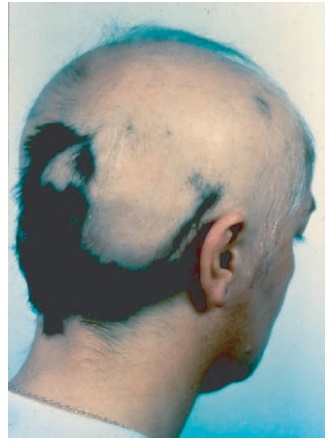
Figure 10.2 The patient has nonscarring alopecia areata covering >90 percent of his scalp. White tufts of hair near the temples are evidence of hair regrowth during active inflammation in the hair bulb, which inhibits pigment transfer from melanocytes to keratinocytes and hair.
Normal
hair growth cycle can be broken down into three phases (1) anagen growth phase
lasting three or more years, (2) cata-gen transitional period lasting two to
four weeks, and (3) telogen phase when the hair follicle advances from the
inferior segment of the follicular sheath into the isthmus and is eventually
shed, either by traction or from being pushed out by a new hair in anagen
phase. Hairs affected by AA end the ana-gen phase prematurely and enter into
telo-gen, resulting in precipitous hair shedding. Because AA does not result in
destruction/ scarring of the hair follicle, “lost” hairs may eventually grow
back, first appearing as “exclamation point” hairs along the border of focal hair loss. These pathognomonic
exclamation point hairs are broader at their distal ends, hence the name.
Although
the exact mechanism of path-ological events is still unknown, there is a
growing body of evidence indicating that it is a T-cell-mediated autoimmune
disease as follows:
1.
The mononuclear infiltrate
surrounding the hair follicle is primarily composed of CD4 and CD8+
T cells as well as mac-rophages.
2. Type I T
cells produce IFN-γ, which
is associated with increased expression of HLA-DR, HLA-ABC, and ICAM-1 in the
follicular epithelium, resulting in increased leukocyte trafficking from blood
into the hair follicle.
3. Hair
regrowth is reproducibly induced by immunosuppressive drug treat-ment,
including local corticosteroid injections and the use of systemic cyclosporine.
4.
Lesional scalp from AA patients
grafted onto SCID mice regrows, coinciding with the loss of the infiltrating
lympho-cytes in the graft. Furthermore, hair loss can be transferred to human
scalp explants in SCID mice by injection of lesional T cells.
Circulating
autoantibodies to follicular structures have been reported in biopsies of AA
patients but have also been reported in normal controls. These autoantibodies
have also been seen in C3H/HeJ mice and DEBR rats but do not appear pathogenic
in either mice or humans. Also one cannot transfer AA by injection of patient
IgG into human skin explants.
In
contrast, C3H/HeJ mice develop spontaneous hair loss with aging and
dem-onstrate many features of AA, including inflammatory infiltrates and
response to intralesional steroids. More important, grafts from C3H/HeJ mice
when implanted into C3H/SmNCPrkd (SCID/J) mice do not result in hair loss,
which emphasizes the role of the host immune system. In common with human AA,
there is elevated expression of MHC class I and II antigens and ICAM-1 in the
skin of these animals, and they respond to topical immunother-apy like humans.
As in
many other autoimmune dis-eases, there is a genetic susceptibility to the
disease. AA has HLA associations with DQB/*03 and possibly HLA-A. These associations
suggest a role for CD4+ T cells in AA but association with HLA, ABC
was not examined in these studies, thus excluding the possible role of CD8+
T cells. Autoimmune diseases result from many factors, and it is probable that
AA is polygenic with multiple potential routes of genetic susceptibility. In
addition to HLA genes, there is known genetic polymor-phism in cytokine
receptors and antigen-processing molecules. Thus, genetics alone is unlikely to
supply a complete explana-tion of disease development.
If there
is a T-cell-mediated compo-nent to this disease coupled with a genetic
susceptibility, what antigens stimulate this T-cell activation? Among the
recent hypothesis has been the suggestion that the autoantigen in AA is the
melanocyte. Evidence that supports this conclusion includes the clinical
observation that with disease activity pigmented hairs are lost more quickly
then nonpigmented/white hairs. Second, melanocytes are a significant component
of the hair bulb, which is the site of the immunological attack. Finally, there
is an association of AA with vitiligo – a disease of focal melanocyte ablation.
Sup-portive evidence can also be found in the animal models. Using SCID mice
with skin grafts obtained from AA patients, mela-nocyte-associated peptides are
capable of activating lesional T cells to induce hair loss. However, melanocyte
antigens may not be the only autoantigens capable of stimulating these cells.
Treatment
at present is related to the observation that AA appears to be medi-ated by a TH1
response with production of IFN-γ.
Psoriasis also has a TH1 response. IL-10, which inhibits TH1
responses, was found to be effective in psoriasis in a phase II clinical trial.
On the basis of these results and a small trial in AA, the use of IL-10 in
intralesional AA might be warranted. The same might be said of alefacept and
mono-clonal antibodies that are directed against CD11a (LFA-1) or CD4 cells.
All of these immunomodalities are presently available and could be considered
as potential new therapeutics if an appropriate risk-to-bene-fit equation is
established in clinical trials.
Related Topics