Chapter: Medical Immunology: Immune Complex Diseases
Physiopathology of Immune Complex Disease
PHYSIOPATHOLOGY OF IMMUNE COMPLEX DISEASE
The formation of an immune complex does not have direct pathological consequences. The pathogenic consequences of immune complex formation depend on the ability of those immune complexes to:
· Form a stable bond with a circulating cell or to leave the intravascular compartment and become tissue-fixed
· Activate the complement system
· Interact with cells able to release enzymes and mediators involved in inflammation
All of these properties are related to the physicochemical characteristics of immune com-plexes (Table 23.1).

A. Physicochemical Characteristics of Immune Complexes
Size, affinity of the Ag.Ab reaction, and class and subclass of antibodies involved in im-mune complex formation are among the most important determinants of the pathogenic sig-nificance of IC.
Very large Ag.Ab aggregates containing IgG1 or IgG3 antibodies will activate com-plement very effectively but are usually nonpathogenic. This is due to a combination of facts: very avid ingestion and degradation by phagocytic cells and difficulty in diffusing across the endothelial barrier. In contrast, very small complexes (Ag1.Ab1-3), even when involving IgG1 and IgG3 antibodies, are able to diffuse easily into the extravascular com-partment, but are usually nonpathogenic because of their inability to activate complement. Actually, the most potentially pathogenic IC are those of intermediate size (Ag2-3.Ab2-6), particularly when involving complement-fixing antibodies (IgG1, IgG3) of moderate to high affinity. However, a main question that needs to be answered is how these IC diffuse into the subendothelial space.
B. Immune Complex Formation and Cell Interactions
1. Circulating Immune Complexes
IC formed in the circulation may be deposited in various tissues, where they cause inflam-mation and tissue damage. Characteristically, multiple organs and tissues may be affected and the clinical paradigms are diseases such as serum sickness and systemic lupus erythe-matosus. The interaction of IC with cells able to release mediators of inflammation appears to be considerably enhanced if the IC are surface-bound, rather than soluble. This immobi-lization of immune complexes along vessel walls is likely to be mediated by:
1. C3 receptors, such as those located on the glomerular epithelium
2. C1q receptors, expressed by endothelial cells
3. Fc receptors, expressed on the renal interstitium and by damaged endothelium
4. Affinity of some antigen moieties in IC for specific tissues, such as that of DNA for glomerular basement membrane and collagen
2. Formation of Immune Complexes In Situ
Direct injection of antigen into a tissue will result in local IC formation with circulating an-tibody. Examples include the Arthus reaction (antigen injected into the dermis binds circu-lating antibody) and hypersensitivity pneumonitis (antigen inhaled forms IC with circulat-ing antibody).
Other types of IC formed in situ include those formed when antibodies react with antigens present on the cell surface membrane of circulating or tissue cells. Immune com-plexes formed on cell membranes can lead to the destruction of the cell, either by promot-ing phagocytosis or by causing complement-mediated lysis. This mechanism is responsi-ble for the development of various immune cytopenias.
Autoantibodies may also bind to basement membrane antigens, as in Goodpasture’s syndrome, or may react with an antigen that has become adsorbed to a basement membrane due to charge interactions, such as seems to be the case of DNA, which binds to basement membrane collagen. The formation of IC in synovial membranes may also result from a two-step in situ reaction between monomeric, freely diffusible autoantibodies and synovial membrane antigens.
3. Adsorption and Transfer of Immune Complexes
Circulating IC can bind to platelets and red cells. Human platelets express Fc receptors, specific for all IgG subclasses and CR4, which binds the C3dg fragment of C3. Red blood cells (RBC) express CR1, through which C3b-containing IC can be bound. In addition, IC can bind to RBC through nonspecific interactions of low affinity, which do not require the presence of complement. Immune complex binding to RBC is believed to be an important mechanism for clearance of soluble IC from the systemic circulation. Experimental work in primates and metabolic studies of labeled IC in humans show that RBC-bound IC are maintained in the intravascular compartment until they reach the liver, where they are pre-sented to phagocytic cells. The phagocytic cells have Fc receptors able to bind the IC with greater affinity than the red cells; as a consequence, the IC are removed from the RBC membrane, while the red cells remain undamaged (Fig. 23.2).
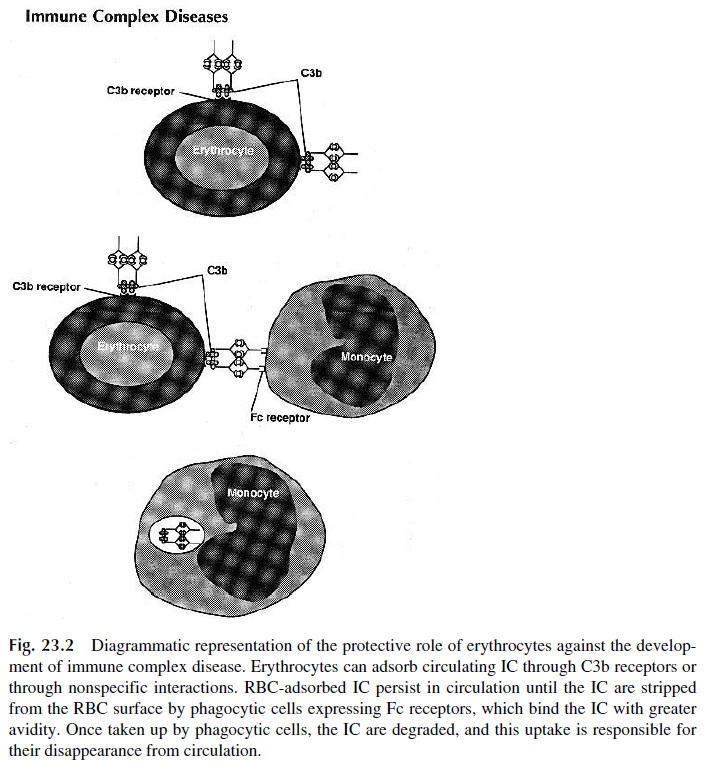
C. Tissue Deposition of IC
Our understanding of how immune complexes induce inflammation and tissue damage is still very incomplete, particularly in cases where the tissue damage is believed to result from the extravascular deposition of circulating immune complexes. A major obstacle to such deposition is the endothelial barrier, which is poorly permeable even to intermediate-sized immune complexes. How this barrier is overcome remains an unanswered question. Another question that has not been fully answered is what determines the preferential de-position of IC in specific tissues.
Deposited IC are frequently found around the small vessels of the skin, particularly in the lower limbs, the kidney glomeruli, the choroid plexus, and the joints. It is likely that regional factors may influence the selectivity of IC deposition. For example, the preferen-tial involvement of the lower limbs in IC-related skin vasculitis may result from the simple fact that the circulation is slowest and the hydrostatic pressure highest in the lower limbs. On the other hand, The frequent involvement of the kidney in IC-associated disease may be a consequence of the existence of C3b receptors in the renal epithelial cells, Fc recep-tors in the renal interstitium, and a collagen-rich structure (the basement membrane), which can also be involved in nonspecific interactions.
In some cases, in situ deposition of IC may be a two-step process: first the antigen may diffuse across the endothelium and become associated with extravascular structures. Monomeric antibody, equally diffusible, may later become associated with the immobi-lized antigen. The best example for this sequence is SLE-associated nephritis: DNA becomes associated with the glomerular basement membrane, and anti-DNA antibodies cross the endothelial barrier and become associated with DNA at the basement membrane level.
Any pathogenic sequence involving the deposition of circulating IC has to take into account increased vascular permeability in the microcirculation, allowing the diffusion of small- to medium-sized soluble IC to the subendothelial spaces (Fig. 23.3). The initial step is likely to be the activation of monocytes or granulocytes by immobilized IC, resulting in the release of vasoactive amines and cytokines. Receptor-mediated interactions involving Fc receptors or complement receptors on endothelial cells could play the initiating role by immobilizing IC at the level of the microvasculature. The retention of soluble IC diffusing through the endothelium in the kidney could be mediated by C3b receptors of the renal ep-ithelial cells or by Fc receptors in the renal interstitium. The mechanisms involved in se-lective retention of soluble IC in other tissues, such as the skin and choroid plexus, are poorly understood.
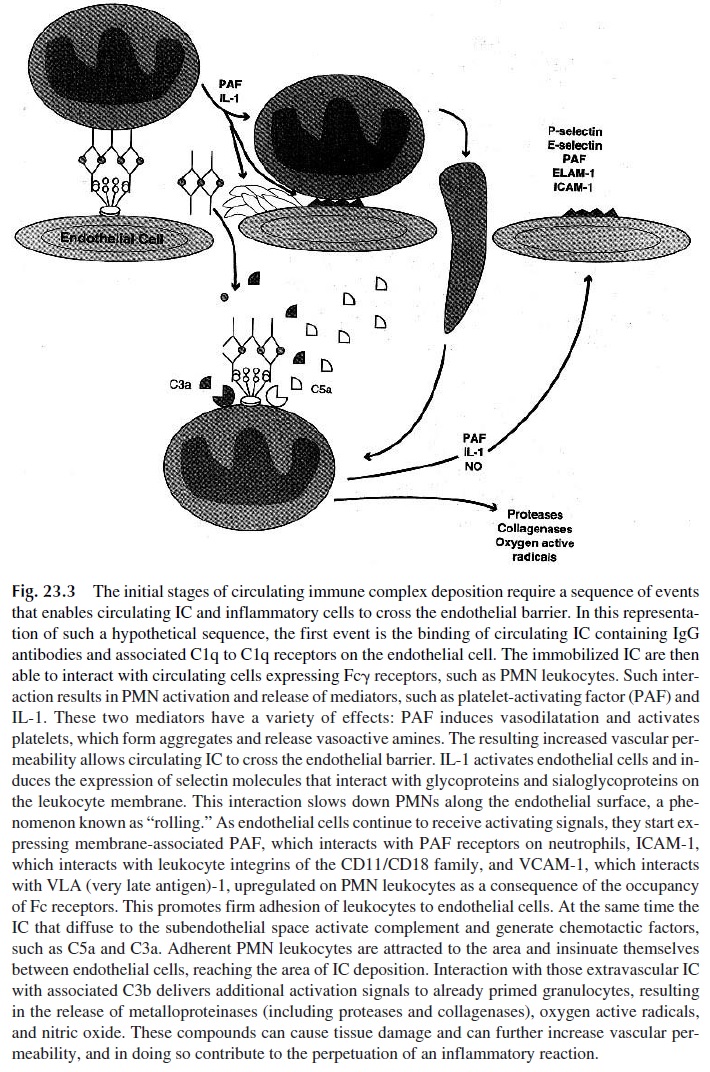
D. Inflammatory Circuits Triggered by Immune Complexes
The development of inflammatory changes after extravascular formation or deposition of immune complexes is not observed in experimental animals depleted of neutrophils or complement. Activated macrophages and the soluble compounds released as a conse-quence of their activation also play important roles (Table 23.2).

Complement components play a significant role as opsonins and chemotactic factors, while activated granulocytes can release a wide variety of proteolytic enzymes that medi-ate tissue damage.
IC containing IgM, IgG1 and/or IgG3 antibodies, able to activate the complement system by the classical pathway, are believed to have the greatest pathogenic potential. Ac-tivation of the complement cascade results in the generation of chemotactic and pro-in-flammatory fragments, such as C3a and C5a . These complement compo-nents have strong pro-inflammatory effects, mediated by a variety of mechanisms:
· C5a increases vascular permeability directly as well as indirectly (by causing the re-lease of histamine and vasoactive amines from basophils and mast cells).
· C5a enhances the expression of the CD11/CD18 complex on neutrophil membranes, increasing their adhesiveness to endothelial cells.
· C5a and C3a are chemotactic for neutrophils, attracting them to the area of IC depo-sition and stimulating their respiratory burst and release of granule con-stituents.
The combination of chemotaxis, increased adherence, and increased vascular perme-ability plays a crucial role in promoting extravascular emigration of leukocytes. It needs to be stressed that the inflammatory process triggered by immune complexes is characteristi-cally associated with extravascular granulocyte infiltrates. The emigration of neutrophils and other granulocytes is regulated by a series of interactions with endothelial cells, known as the adhesion cascade. This cascade involves the following sequence of events:
1. The initial event involves the upregulation of selectins (P-selectin and E-se-lectin) on endothelial cells, which can be caused by a variety of stimuli (e.g., his-tamine, thrombin, bradikinin, leukotriene C4, free oxygen radicals, or cy-tokines). The consequence of this upregulation is the slowing down (rolling) and loose attachment of leukocytes (expressing a third selectin, L-selectin, which binds to membrane oligosaccharides on endothelial cells). These initial interac-tions are unstable and transient.
2. The endothelial cells, in response to persistent activating signals, express platelet-activating factor (PAF) and ICAM-1 on the membrane. Neutrophils ex-press constitutively a PAF receptor that allows rolling cells to interact with mem-brane-bound PAF.
3. The interaction of neutrophils with PAF, as well as signals received in the form of chemotactic cytokines such as IL-8 (which can also be released by endothe-lial cells), activate neutrophils and induce the expression of the following inte-grins: CD11a/CD18, LFA-1, and related molecules, and very late antigen-4, and VLA-4.
4. The interaction between integrins expressed by neutrophils and molecules of the immunoglobulin superfamily expressed by endothelial cells (ICAM-1 and re-lated antigens bind LFA-1 and related molecules; VCAM-1 binds VLA-4) causes firm adhesion (sticking) of inflammatory cells to the endothelial surface, which is an essential step leading to their extravascular migration. VLA-4 is also expressed on the membrane of lymphocytes and monocytes, and its interaction with endothelial VCAM-1 allows the recruitment of these cells to the site of in-flammation.
5. The interactions between integrins and their ligands are important for the development of vasculitic lesions in patients with systemic lupus erythematosus and other systemic autoimmune disorders and of purulent exudates in infection sites. Patients with genetic defect in the expression of CD18 and related cell adhesion molecules (CAMs) fail to form abscesses be-cause their neutrophils do not express these molecules and fail to migrate.
6. The actual transmigration of leukocytes into the subendothelial space seems to in-volve yet another set of CAMs, particularly one member of the immunoglobulin superfamily known as PECAM (platelet endothelial cell adhesion molecule), which is expressed both at sites of intercellular junction and on the membranes of leukocytes. PECAM-1 interacts with itself, and its expression is upregulated on both endothelial cells and leukocytes by a variety of activating signals.
7. The egression of leukocytes from the vessel wall is directed by chemoattractant molecules released into the extravascular space and involves diapedesis through endothelial cell junctions.
As leukocytes begin to reach the site of immune complex formation or deposition, they continue to receive activating signals. Their activation brings about the release of ad-ditional chemotactic factors and continuing upregulation of CAMs on endothelial cells, the efflux of phagocytic cells to the subendothelial space will intensify, and the conditions needed for self-perpetuation of the inflammatory process are created. All polymorphonu-clear leukocytes express Fcγ receptors and C3b receptors that mediate their binding and in-gestion of IC. This process is associated with activation of a variety of functions and with the release of a variety of cytokines, enzymes, and other mediators. One of the mediators released by activated neutrophils is platelet-activating factor, which will promote the self-perpetuation of the inflammatory process by:
· Increasing vascular permeability (directly or as a consequence of the activation of platelets, which release vasoactive amines)
· Inducing the upregulation of the CD11/CD18 complex on neutrophils
· Inducing monocytes to release IL-1 and TNFα , which activate endothelial cells and promote the upregulation of adhesion molecules (E and P selectins) and the synthesis of PAF and IL-8
Furthermore, as granulocytes try to engulf large IC aggregates or immobilized IC, they become activated and release their enzymatic contents, including metalloproteinases with protease and collagenase activity and oxygen active radicals. These compounds can damage cells, digest basement membranes and collagen-rich structures, and contribute to the perpetuation of the inflammatory reaction by causing direct breakdown of C5 and C3 and generating additional C5a and C3b. The formation of C3b promotes activation of the alternative pathway, thus continuing to amplify the pro-inflammatory reaction. As the in-flammatory reaction continues to intensify, clinical manifestations emerge. The clinical manifestations of immune complex disease depend on the intensity of the inflammatory re-action and on the tissue(s) predominantly affected by IC deposition.
Related Topics