Chapter: Medical Immunology: Primary Immunodeficiency Diseases
Phagocytic Cell Deficiencies
PHAGOCYTIC CELL DEFICIENCIES
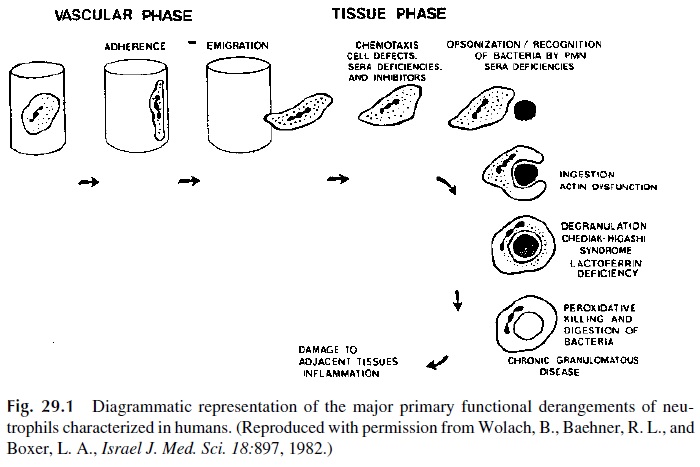
Cell findings in patients with phagocytic disorders include infections of soft tissues, cel-lulitis, and chronic recurrent lymphadenitis. Recurrent abscesses in organs such as lung, liver, and bone are also frequent. Commonly these infections are due to Staphylococcus sp., Pseudomonas sp., Candida sp., and Aspergillus sp. Chronic or recurrent oral ulcers, poorwound healing, periodontal disease, and delayed separation of the umbilical cord are early hallmarks of deficiencies in phagocytic cell function or numbers. Phagocytic function dysfunction can result from either quantitative or qualitative defects in phagocytic cells. Figure 29.1 diagrammatically illustrates the aspects of PMN function that can be affected in different pathological situations.
A. Disorders of Leukocyte Adherence
The most common among these very rare disorders is the lack of expression of the CD11/CD18 complex, which affects phagocytic cells and T lymphocytes. This disease is inherited as an autosomal recessive trait. Initial clinical manifestations include delayed sep-aration of the umbilical cord and chronic leukocytosis. During childhood, these individu-als suffer from repeated pyogenic infections and, with less frequency, fungal infections. Se-vere necrotizing gingivitis and periodontal disease is common.
B. Job’s Syndrome (Hyper-IgE Syndrome)
Chronic severe atopic dermatitis, very high levels of serum IgE, and recurrent staphylo-coccal infections of the lungs and cutaneous abscesses characterize this syndrome. Inheri-tance is often autosomal dominant. Other types of pyogenic infections, particularly of the upper airways, and chronic candidiasis can also be present.
The mechanism of this disease has not been well defined. A defect of monocyte chemotaxis has been reported in most patients, but the severity is quite variable. It is un-likely that this is the primary defect leading to immune deficiency. The high levels of IgE correspond, at least in part, to the production of IgE anti–S. aureus antibodies. In con-trast, IgA antibodies to S. aureus are abnormally low, and other indices of humoral im-mune function (responses to toxoid boosters and to in vitro stimulation with PWM) are also depressed.
C. Chronic Granulomatous Disease
Chronic granulomatous disease (CGD), although rare, is the most frequent primary phago-cytic cell deficiency. The majority of cases are inherited as an X-linked trait, but autosomal recessive inheritance is involved in 25–35% of the cases.
The molecular basis of CGD is heterogeneous. The X-linked form is the result of a mutation in the heavy-chain (91 kDa) phosophoprotein (alphox) of cytochrome B244. Molecular genetic studies have shown that in about half of the cases of X-linked CGD there is a failure to express mRNA for the alphox protein and in the remainder of cases, mRNA is present but there is a failure to transport or properly insert the protein in the cytoplasmic membrane. The remaining cases of CGD are inherited as autosomal recessive traits, in-volving other cytochrome proteins such as P47, p67, or the light chain (22 kDa) of cy-tochrome B.
All of the cytochrome B deficiencies result in a lack of functional oxidase at the cell membrane level. There is a failure to generate superoxide and H2O2, and consequently, in-tracellular killing is defective. Both types of professional phagocytic cells (PMN leuko-cytes and monocytes) are affected.
The killing defect leads to infections with catalase-positive organisms such as Staphylococci, Serratia marscecens, Klebsiella sp., Aerobacter sp., Salmonella sp., Chro-mobacterium violaceum, Pseudomonas cepacia, Nocardia sp., and Aspergillus sp. Cata-lase-negative organisms (such as Streptococcus pneumoniae) do not usually cause infec-tions in these patients. The lack of involvement of catalase-negative organisms results from the fact that when ingested by phagocytic cells they continue to generate H2O2 that they cannot degrade. The H2O2 generated by the bacteria progressively accumulates in the phagosome, eventually reaching bactericidal levels.
The clinical manifestations are dominated by recurrent bacterial and fungal infec-tions. The most frequent infection sites are the lungs, lymph nodes, liver, skin, and soft tis-sues. The infections are characterized by the formation of microabscesses and noncaseat-ing granulomas. Suppurative lymphadenitis, pyoderma, pneumonia with suppurative complications, liver abscesses, osteomyelitis, and severe periodontal disease are among the most frequently seen infections. Generalized lymphadenopathy and hepatosplenomegaly are frequently found on physical examination. Because the infectious agents are often se-questered within leukocytes or walled-off in granulomas, it is common for blood or tissue cultures to be negative. Most patients have elevated levels of the major immunoglobulins (hypergammaglobulinemia).
Diagnosis is usually confirmed by abnormal results in one of the variations of the NBT reduction test .
Infections are treated with antimicrobials chosen on the basis of susceptibility studies if bacteria have been recovered from infection sites. Prophylactic administration of trimethoprim-sulfamethoxazole is generally recommended. Interferon-Îł administration has been found to result in a decrease in the frequency of infectious episodes in some patients with CGD. Early trials suggested that interferon-Îł enhanced the neutrophil respiratory burst and that the effect was more pronounced in patients with the autosomal variants of CGD.
The identification of the genes coding for the different molecular components of the oxidase system has led to trails of gene therapy by using retroviral vectors to insert the gp91 cytochrome B299 gene into the patient’s CD34+ stem cells. It is hoped that when these cells are returned to the patient it will result in the emergence of oxidase-positive phago-cytic cells.
D. Chédiak-Higashi Syndrome
This rare disease is due to abnormalities in the cytoplasmic granules involved in the for-mation of the phagosome leading to impaired killing of certain microorganisms. It is be-lieved that the primary defect may be in the regulation of membrane activation. The PMN leukocytes are able to ingest microorganisms, but the cytoplasmic granules tend to coalesce into giant secondary lysosomes, with reduced enzymatic contents, that are inconsistently delivered to the phagosome. As a consequence, intracellular killing is slow and inefficient. NK cell function has also been reported to be impaired.
1. Clinical Manifestations
Mucocutaneous albinism, recurrent neutropenia, unexplained fever, and peripheral neu-ropathy characterize patients with this syndrome. Many patients develop hep-atosplenomegaly and lymphadenopathy associated with recurrent bacterial and viral infec-tion, fever, and prostration. End-stage disease is associated with a poor prognosis.
2. Diagnosis
The finding of morphological abnormalities in PMN leukocytes (giant lysosomes) supports the diagnosis. Phagocytic cells killing tests show impaired intracellular killing.
3. Treatment
Infections are treated symptomatically with antibiotics. Ascorbic acid administration has been shown to increase bactericidal activity in some patients. This improvement may be re-lated to an effect of ascorbic acid on membrane fluidity, which is abnormally high in the patient’s PMN leukocytes and is normalized by ascorbic acid.
Related Topics