Chapter: Medical Immunology: Primary Immunodeficiency Diseases
Complement Deficiencies - Primary Immunodeficiency Diseases
COMPLEMENT DEFICIENCIES
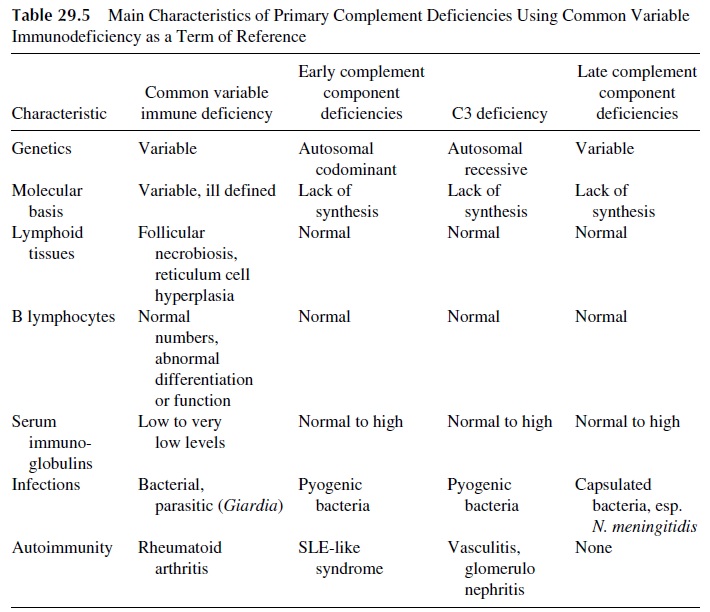
Deficiencies of virtually all components of the complement cascade have been reported. Clinical findings associated with complement deficiencies are highly variable and dependent on the location of the defect within the complement cascade. Symptoms range from an increased risk for the development of autoimmune disease to recurrent infections. The deficiencies can be grouped as early-component (C1, C2, and C4) deficiencies, late-com-ponent (C5 to C9) deficiencies, C3 deficiency, and defects in the alternative complement cascade. The characteristics of these groups are summarized in Table 29.5.
A. Early Component Deficiencies
Deficiencies of C1, C2, and C4 can be asymptomatic, can be associated with predisposition to infections, or, most frequently, can be associated with clinical symptoms suggestive of autoimmune disease. C2 and C4 deficiencies are inherited as an autosomal co-dominant trait and are often associated with a syndrome mimicking systemic lupus erythematosus (SLE), although the clinical evolution is more benign and renal dysfunction is less frequent. Patients with homozygous C2 or C4 deficiency often have associated defects in factor B. Recurrent viral and bacterial infections are common. Persistent levels of circulating im-mune complexes are often found in patients with C2 or C4 deficiency, probably as a result of altered dynamics of immune complex clearance. As a result of low levels of C2 or C4 and due to the interruption in the activation sequence, these patients do not generate nor-mal amounts of C3b.
B. C3 Deficiency
Primary C3 deficiency is a rare condition, transmitted as an autosomic recessive trait. Pa-tients with C3 deficiency have an inability to opsonize antigens and suffer from recurrent pyogenic infections from early in life, with a clinical picture similar to that of X-linked in-fantile agammaglobulinemia with normal B- and T-cell function. This is not surprising, given the pivotal role played by C3 in complement activation and in opsonization of mi-crobial agents. Furthermore, C3-deficient patients may present manifestations of immune complex disease, such as glomerulonephritis and vasculitis.
Factor I (C3b inactivator) and factor H deficiencies result in a deficiency of C3 sec-ondary to a fourfold increase in the catabolic rate of this complement component. Patients are prone to recurrent pyogenic infections, particularly with encapsulated bacteria such as S. pneumoniae and N. meningitidis. They are also prone to the development of immunecomplex disease. “Anaphylactoid” reactions secondary to the spontaneous generation of C3a are also frequently observed in these patients.
C. Late Component Deficiencies
Deficiencies of C5, C6, C7, C8, and C9, complement compounds that make up the mem-brane attack complex, have been reported to be associated with increased frequency of in-fections, most commonly due to bacteria with polysaccharide-rich capsules such as Neis-seria sp.
Case 29.1 Revisited
The prevalence of bacterial infections (pneumonia, pyoderma) caused by classic pyogenic organisms (e.g., Haemophilus influenzae) is strongly suggestive of humoral immunodefi-ciency. The lack of adverse reactions after the usual childhood immunizations, which in-clude several attenuated viruses, and the uneventful course of chickenpox strongly indi-cates that cell-mediated immunity is probably normal.
The clinical spectrum of the humoral immunity deficiency, apparent integrity of cell-mediated immunity, normal numbers of B lymphocytes, and age of onset (in later child-hood) points to a diagnosis of variable immunodeficiency. The pattern of inheritance is variable but in this case is most likely autosomal recessive. Male and female children have an equal chance of carrying the same disease. Diagnosis would be impossible in the neona-tal and early infancy period, since any child born to this mother would be severely hy-pogammaglobulinemic and likely to suffer from bacterial infections. Also, the onset of dis-ease could be delayed for several years.
Two types of tests could be valuable in this patient: the assay of anti–H. influenzae antibodies (was negative), and a lymphocyte transformation assay, measuring IL-2 release after stimulation with PHA and anti-CD3 monoclonal antibodies. Patients with common variable immunodeficiency often show abnormal mitogenic responses, low gp39 expres-sion, and subnormal IL-2 release after T-cell mitogenic stimulation, suggesting that the ba-sis of the disease is lack of T-cell help.
As in infantile agammaglobulinemia, chronic obstructive lung disease and bronchiectasis are likely to develop as a consequence of having repeated bronchopul-monary infections.
The best treatment for this condition is the administration of intravenous gamma globulin. The periodicity of administration needs to be established for each individual pa-tient, based on the longitudinal variation in serum immunoglobulin levels and on the du-ration of the symptom-free period after gamma globulin administration. In most cases, ad-ministrations of IV gamma globulin need to be repeated every 3– 4 weeks.
Related Topics