Chapter: Medical Immunology: Systemic Lupus Erythematosus
Pathogenesis of SLE(Systemic Lupus Erythematosus)
PATHOGENESIS OF SLE
Multiple environmental, hormonal, genetic, and immunoregulatory factors are involved in the expression of the disease. In any given patient, different factors contribute variably to the expression of the disease.
A. Genetic Factors
The understanding of the pathogenic mechanisms underlying the progression of SLE has been facilitated by the discovery of spontaneously occurring disease in mice that resembles SLE in many respects. During the inbreeding of mice, it was observed that the F1 (first-gen-eration) hybrids obtained by mating black and white mice from New Zealand [(NZB×NZW) F1] spontaneously developed a systemic autoimmune disease involving a variety of organs and systems. Throughout the course of their disease, the mice develop hypergam-maglobulinemia, reflecting a state of hyperactivation of the humoral immune system. The animals have a variety of autoantibodies and manifestations of autoimmune disease and im-mune complex disease similar to those seen in humans with SLE. As the disease pro-gresses, they develop nephritis and lymphoproliferative disorders and die.
The importance of genetic factors in the development of disease in NZB mice is un-derlined by the observation that the parental NZB mice have a mild form of the disease manifested by autoimmune hemolytic anemia, but that the introduction of the NZW genetic background made the disease accelerate and worsen. Genetic linkage studies and mi-crosatelite gene marker analysis indicate that many of the immunological abnormalities are under multigenic control, one gene(s) controlling the animal’s ability to produce anti-DNA antibodies, another the presence of antierythrocyte antibodies, and still other genes con-trolling high levels of IgM production and lymphocytic proliferation.
Two other mouse strains that develop a SLE-like disease spontaneously have been identified: MRL lpr/lpr and MRL gld. The first strain has a defect in the Fas gene, whereas the second has a defect in the Fas ligand gene. The products of these two genes are re-sponsible for the programmed cell death of cells also known as apoptosis, which is critical for the control of undesirable immune responses. Only rare patients with lupus have struc-tural defects of the Fas/Fas ligand proteins.
Several pieces of evidence indicate that genetic factors also play a role in the patho-genesis of human SLE. Serum DNA and T-cell antibodies as well as cellular abnormalities are present in healthy relatives of lupus patients. There is moderate degree of clinical dis-ease concordance among monozygotic twins. The fact that the clinical concordance be-tween twins is only moderate strongly indicates that genetic factors alone may not lead to the expression of the disease and that other factors are needed. The genes that could play a role, probably in synergy with environmental factors, have not been identified. Current ev-idence indicates that in humans, as in mice, these genes are probably linked to the MHC. For example, the HLA-DR2 haplotype is overrepresented in patients with SLE. Also, as mentioned before, an SLE-like disease develops frequently in individuals with C4 and C2 deficiencies (C4 and C2 genes are located in chromosome 6, in close proximity to the MHC genes). Also, individuals lacking C1q are also prone in developing lupus. Recently, genome-wide searches for “lupus” genes have been undertaken. These studies have re-ported various genome areas to be associated with lupus. Interestingly, many of these areas are found in 6p and 1q.
B. Immune Response Abnormalities
SLE is a disease associated with profound immunoregulatory abnormalities, affecting both humoral and cell-mediated responses.
B-Cell Abnormalities
Increased numbers of B cells and plasma cells are detected in the bone marrow and pe-ripheral lymphoid tissues secreting immunoglobulins spontaneously. The number of these cells correlates with disease activity. Only a limited number of light and heavy-chain genes are used by autoantibodies, demonstrating that the autoantibody response involves only a few of all B-cell clones available. Furthermore, the changes appearing in their sequence over time strongly suggest that they undergo affinity maturation, a process that requires T-cell help. It also suggests that a few antigens drive the response. Immunosuppressive drug treatment of both murine and human lupus causes clinical improvement associated with de-creased B-cell activity. Any infection that induces B-cell activation is likely to cause a clin-ical relapse in patients with inactive SLE.
T-Cell Abnormalities
From our knowledge of the biology of the immune response, it can be assumed that the pro-duction of high titers of IgG anti-dsDNA antibodies in patients with SLE must depend upon excessive T-cell help and/or insufficient control by suppressor T cells. Support for this the-ory is provided by the following observations:
1. In both human and murine lupus, a new subset of CD3+ cells that express nei-ther CD4 nor CD8 has been found to provide help to autologous B cells synthe-sizing DNA antibodies.
2. The finding of anti–T-cell antibodies in the serum of (NZB×NZW) F1 mice and in the sera of humans with SLE raised the possibility that the deletion of a spe-cific subset of regulatory cells could contribute to the inordinate B-cell activity associated with the development of autoimmunity. Obviously, defective sup-pressor T-cell function could enhance the helper T-cell–mediated B-cell overactivity.
3. In humans, anti–T-cell antibodies are also responsible for the lymphopenia that is frequently seen in patients with SLE. This lymphopenia is often associated with findings suggestive of a generalized depression of cell-mediated immunity, such as decreased lymphokine production (IL-1 and IL-2) and lack of reactivity (anergy) both in vivo and in vitro to common recall antigens, particularly during active phases of human SLE. The impairment of cell-mediated immunity may explain the increased risk of severe opportunistic infections in patients with SLE.
4. Extensive deletions in the T-cell repertoire have been found in NZW mice in which the C β2 and D β2 genes of the T-cell antigen receptor are missing. These deletions could be associated with a faulty establishment of tolerance to self-MHC during intrathymic ontogeny.
5. In humans, restriction fragment length polymorphism studies of the constant re- gion of the TcR demonstrated an association between TcRα chain polymor-phism and SLE and TcR chain polymorphism and production of anti-Ro anti-bodies. More recently, sequence information of the TCRβ chains of pathogenic human T-cell clones demonstrated bias in the T-cell repertoire selection process, whose meaning is still to be defined. The immune response is thus “oligoclonal” in both T and B compartments.
C. Immune Complexes in SLE
The pathogenic role of immune complexes (IC) in SLE has been well established. As sum-marized in Figure 18.1, this pathogenic role is a result of a variety of abnormal circumstances.
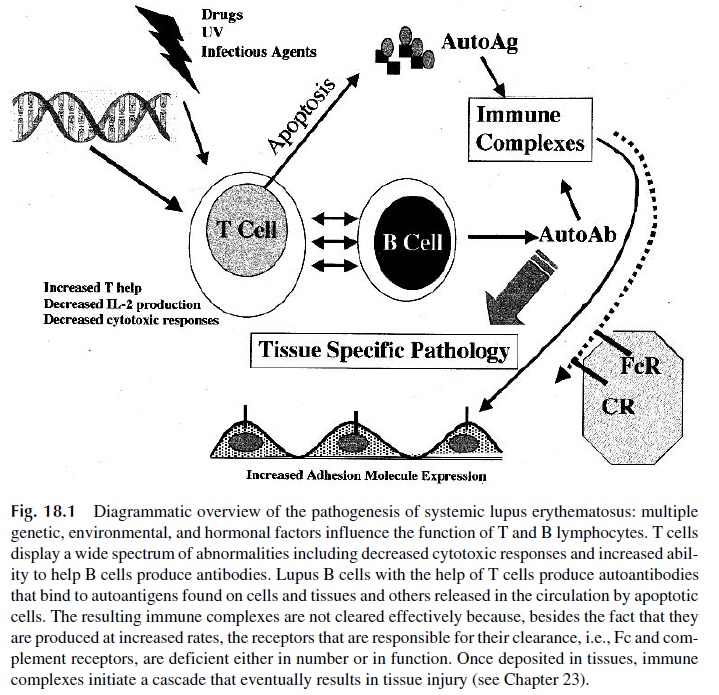
First, marked elevations in the levels of circulating immune complexes can be de-tected in patients with SLE sera during acute episodes of the disease by a variety of tech-niques . Since patients with active SLE have high levels of free circulating DNA and most also have DNA antibodies, DNA–anti-DNA IC are likely to be formed ei-ther in circulation or in collagen-rich tissues and structures such as the glomerular basement membrane, which have avidity for DNA.
Besides the fact that immune complexes are formed at increased rates in patients with SLE, the clearance rate of circulating immune complexes is decreased. Several factors seem to contribute to the impaired clearance of immune complexes:
1. Immune complexes are cleared by the Fc receptor bearing cells of the reticu-loendothelial system. This function has been found to be decreased in patients with SLE. Many patients with lupus nephritis have alleles of Fc receptors that bind IgG with less avidity.
2. Immune complexes often have adsorbed complement components and split products, including C3b, which reacts with CR1. Consequently, IC are trans-ported to the reticuloendothelial system by red blood cells, which bind them through their CR1. Patients with SLE have decreased numbers of CR1, a fact that may compromise the clearance of immune complexes and contribute to the de-velopment of IC-induced inflammatory reactions.
3. Immune complexes are partially solubilized as a consequence of complement activation, a process that contributes to their inactivation and clearance. Individ-uals with C4 deficiency develop a disease with clinical features resembling those of SLE. This observation can be explained by the fact that immune complexes are cleared at slower rates in C4-deficient individuals, perhaps due to the role of C4 fragments in the solubilization and clearance of circulating immune complexes.
The pro-inflammatory properties of immune complexes in SLE are suggested by a variety of observations. First, rising levels of DNA antibodies in conjunction with falling serum C3 levels (reflecting consumption by antigen-antibody complexes) are associated with disease flares.
Second, patients with IgG1 and IgG3 (complement fixing) DNA anti-bodies develop lupus nephritis more frequently than do patients in whom DNA antibodies are of other isotypes. Glomerulonephritis, cutaneous vasculitis, arthritis, and some of the neurological manifestations of SLE are fully explainable by the development of local in-flammatory lesions secondary to the formation or deposition of IC. What remains unclear is whether tissue-fixed IC are circulating IC that eventually become deposited in tissues or if they result from the formation of antigen-antibody complexes in situ.
In SLE patients, immune complex deposits have also been noted on the dermo-epi-dermal junction of both inflamed skin and normal skin, appearing as a fluorescent “band” when a skin biopsy is studied by immunofluorescence with anti-sera to immunoglobulins and complement components (band test).
D. Glomerulonephritis
Immunofluorescence studies indicate that the capillary tufts of renal glomeruli in patients with lupus nephritis contain deposits of immunoglobulins and complement. Several lines of evidence support the conclusion that those deposits represent immune complexes and that these IC are likely to play a primary pathogenic role.
Elution studies have shown that DNA and DNA antibodies are present in these de-posits, confirming that they correspond to antigen-antibody complexes. The deposition of IC in the glomerular basement membrane can be explained in three different ways:
1. Deposition of soluble, circulating IC.
2. Formation of immune complexes in situ. DNA has affinity to glomerular base-ment membranes and, once immobilized, may react with circulating DNA anti-bodies to form antigen-antibody complexes.
3. Cross-reaction of DNA antibodies with collagen and cytoskeleton proteins.
Currently in situ formation of DNA–anti-DNA immune complexes appears as the most likely initiating event, but regardless of how they are formed, IC in the basement membrane are considered nephritogenic because they may activate complement and cause inflammation.
The pathogenic role of IC deposited in the kidney is supported by the fact that there is ample evidence for complement activation via the classic and the alternative pathway in patients with active nephritis. Circulating levels of C3 and C4 are usually decreased, whereas plasma levels of complement breakdown products such as C3a, C3d, and Bb are increased. C1q, C3b, and complement split products such as C3d, C3bi, and C3c can be de-tected attached to circulating immune complexes.
E. Nonimmune Factors Influencing the Course of SLE
In addition and in close interplay with genetic and immunological factors, a variety of other factors have an apparent effect on the evolution of the disease.
1. Hormonal Effects
The expression of the genetic and immunological abnormalities characteristic of murine lupus-like disease is influenced by female sex hormones. For example, in (NZBxNZW) F1 mice, the disease is more severe in females. Administration of estrogens aggravates the evolution of the disease, which is only seen in castrated male mice and not in complete males.
The extent of the hormonal involvement in human SLE cannot be proven directly, but the large female predominance (9:1 female-to-male ratio) as well as the influence of pu-berty and pregnancies at the onset of the disease, or the severity of the disease’s manifes-tations, indicates that sex hormones play a role in the modulation of the disease. Recent in-vestigations suggest that estradiol causes an increased expression of calcineurin, which could lead to increased synthesis of pro-inflammatory cytokines, particularly of the TH2 group. This could lead to an exaggeration of humoral immunity abnormalities during preg-nancy, which in turn could exacerbate the inflammatory processes secondary to IC forma-tion.
2. Environmental Factors
Several environmental insults have been related to the onset or relapse of SLE. Sunlight ex-posure was the first environmental factor influencing the clinical evolution of human SLE to be identified. Exposure to sunlight may precede the clinical expression of the disease or disease relapse. This could be related to the fact that the Langerhans cells of the skin and keratinocytes release significant amounts of interleukin-1 upon exposure to UV light and could thus represent the initial stimulus tipping off a precarious balance of the immune system.
Infections also seem to play a role. The normal immune response to bacterial and vi-ral infections may spin off into a state of B-cell hyperactivity, triggering a relapse.
Drugs, particularly those with DNA-binding ability, such as hydantoin, isoniazide, and hydralazine, can cause a drug-induced lupus-like syndrome. These drugs are known to cause DNA hypomethylation. Because hypomethylated genes are transcribed at higher rates, it is theoretically possible that they cause SLE by increasing the transcription rate of genes involved in the expression of the disease. ANA antibodies appear in 15–70% of pa-tients treated with any of these drugs for several weeks. These ANA antibodies belong, in most cases, to the IgM class and react with histones. Only when the antibodies switch from IgM to IgG does the patient become symptomatic. These ANA usually disappear after ter-mination of the treatment. Patients with drug-induced SLE usually have a milder disease, without significant vital organ involvement.
Case 18.1 Revisited
The patient’s fatigue could just be a reflection of a systemic inflammatory disease but could also be due to anemia. Hemolytic anemia is not infrequent in SLE. Seizures and other neu-rological symptoms may be due to (1) deposition of immune complexes in CNS tissues, (2) binding of antineuronal autoantibodies, with or without complement activation, (3) the ef-fect of infiltrating autoreactive T cells, or (4) the effects of neurotoxins (such as quinolinic acid) released by activated immune cells.
Patients with SLE often develop antiphospholipid antibodies. Although the exact pathogenic sequence is not known, these antibodies interfere with clotting factors, causing vascular thrombosis usually without vasculitis.
The pathogenesis of skin lesions is likely to involve several factors. Deposition of IC at the dermo-epidermal junction is likely to play a role, but not the only one, since this de-position can be observed in normal skin. Exposure to sunlight is likely to play a significant role as well, perhaps because the Langerhans cells and keratinocytes of the skin release significant amounts of interleukin-1 upon exposure to UV light. This could represent an ad-ditional trigger to a local inflammatory reaction, involving both the effects of UV exposure and the effects of IC deposition.
In this patient, several complications would cause concern: autoimmune hemolytic anemia may be extremely difficult to treat, and the progression of her CNS involvement would also raise considerable therapeutic problems. In addition, deterioration of kidney function due to the development of lupus nephritis is always a major concern in any patient with SLE.
This patient presented with several clinical features typical of SLE: facial erythema, arthritis, seizures, and possible anemia. The detection of autoantibodies such as those di-rected against dsDNA or the Sm antigen would be confirmatory, but even if only nonspe-cific antinuclear antibodies were detected, a diagnosis of SLE should still be entertained. To evaluate the cause of some of the most striking symptoms of this patient, it would be in-dicated to perform a complete blood count, and if anemia was present, Coombs’ tests should be ordered. Other important tests to be ordered include x-rays of the hands, rheumatoid factor, anticardiolipin and antiphospholipid antibodies, serum creatinine, and urine protein (to evaluate kidney function).
One of the most difficult questions to answer is what triggers the onset of an autoim-mune disease. This patient fits in the age and sex group in which SLE is more prevalent. In addition to hormonal influences, it is likely that this patient carries a genetic predisposi-tion to develop SLE (although the precise marker and nature of such predisposition are not yet clearly identified). Most unclear of all is what was the initial stimulus. An infection leading to cross-reactive autoimmunity is a very appealing hypothesis, but we have no clue about the nature of such infection.
Related Topics