Chapter: Modern Pharmacology with Clinical Applications: Metabolism and Excretion of Drugs
Oxidative and Reductive Enzymes: Phase I Reactions
OXIDATIVE AND
REDUCTIVE ENZYMES: PHASE I REACTIONS
Phase I enzymes act by
causing the drug molecule to undergo oxidation or more rarely, reduction.
Examples of oxidation reactions carried out by phase I enzymes are listed in
Table 4.1 and encompass a broad range of drugs with varying chemical
structures. However, as dis-cussed later, there is still a great deal of
substrate speci-ficity within a given enzyme family.

Cytochrome P450 Enzymes
The cytochrome P450 (CYP450)
enzyme superfamily is the primary phase I enzyme system involved in the
ox-idative metabolism of drugs and other chemicals. These enzymes also are
responsible for all or part of the me-tabolism and synthesis of a number of endogenous
com-pounds, such as steroid hormones and prostaglandins.
Though it was originally
described as the CYP450 en-zyme, it is now apparent that it is a group of
related en-zymes, each with its own substrate specificity. To date, 12 unique
isoforms (e.g., CYP3A4, CYP2D6) have been identified as playing a role in human
drug metabolism, and others may be discovered. These isoforms, along with
examples of compounds for which each isoform plays a substantial role in their
metabolism, are listed in Table 4.2. More than one CYP isoform may be involved
in the me-tabolism of a particular drug. For example, the calcium channel
blocking drug verapamil is primarily metabolized by CYP3A4, but CYPs 2C9, 2C8
and 2D6 participate to some degree, particularly in the secondary metabolism of
the verapamil metabolites. Thus, the degree to which a drug interaction
involving competition for a CYP isoform may occur will depend on the extent of
metabolism of each compound that can be attributed to that isoform. The more
isoforms involved in the metabolism of a drug, the less likely is a clinically
significant drug interaction.

Substrate Specificity of the CYP Enzymes
CYP3A4 is thought to be the most predominant CYP isoform involved in human drug metabolism, both in terms of the amount of enzyme in the liver and the vari-ety of drugs that are substrates for this enzyme isoform.
This isoform may account for
more than 50% of all CYP-mediated drug oxidation reactions, and CYP3A4 is
likely to be involved in the greatest number of drug–drug interactions. The
active site of CYP3A4 is thought to be large relative to other isoforms, as
evi-denced by its ability to accept substrates up to a molec-ular weight of
1200 (e.g., cyclosporine). This active site size allows drugs with substantial
variation in molecular structure to bind within the active site. However, the
fact that two drugs are metabolized predominantly by CYP3A4 does not mean that
coadministration will re-sult in a drug–drug interaction, since drugs can bind
in different regions of the CYP3A4 active site, and these binding regions may
be distinct. In fact, it is believed that two drugs (substrates) can occupy the
active site simul-taneously, with both available for metabolism by the en-zyme.
This finding helps account for a number of absent interactions that would have
been predicted to occur based on strict substrate specificity rules.
CYP3A5, whose amino acid
sequence is similar to that of CYP3A4, appears to possess roughly the same
substrate specificity characteristics as CYP3A4. How-ever, it differs in that
it is not present in all individuals. Thus, patients expressing both CYP3A4 and
CYP3A5 have the potential to exhibit increased metabolism of CYP3A substrates
as compared to individuals express-ing only the CYP3A4 isoform.
Levels of CYP enzyme
expression of any isoform can vary substantially among individuals. The other
identified human CYP3A isoform is CYP3A7, which appears to be expressed only in
the fetus and rapidly disappears following birth, to be replaced by CYP3A4 and
CYP3A5. It is becoming increasingly clear that dif-ferent enzyme expression
patterns, and thus different drug metabolism capabilities, are observed
throughout the various stages of life. Neonates are different from 6-month-old
infants, who differ from year-old infants, who differ from preadolescents, who
differ from adolescents, who differ from adults, who differ from the elderly.
Thus, consideration must be given to the person’s age when assessing drug
metabolism capacity.
The second most common CYP isoform
involved in human drug metabolism is CYP2D6. It may account for 30% of the
CYP-mediated oxidation reactions involv-ing drugs, including the metabolism of
drugs in such diverse therapeutic categories as antipsychotic agents, tricyclic
antidepressants, β-blocking agents, and opioid analgesics. Though this isoform
accepts a number of drugs as substrates, its relative abundance in the liver is
quite low. CYP2D6 is most known for its propensity to exhibit genetic
polymorphisms.
The other isoform responsible for a substantial por-tion (about 10%) of the CYP-mediated drug oxidation reactions is CYP2C9. This
isoform metabolizes several clinically important drugs with narrow therapeutic
in-dices. Two of these drugs are the antiepileptic agent phenytoin and the
anticoagulant warfarin. Any change in the metabolism of these two drugs, either
increased or decreased, can have profound adverse effects. CYP2C9 appears to
prefer weakly acidic drugs as sub-strates, which limits the number of drugs
metabolized by this isoform, since most drugs are weak bases).
The remaining CYP isoforms
involved in human drug metabolism (Table 4.2) are present in the liver in
varying amounts, and each is thought to contribute 2–3% or less of the
CYP-mediated drug oxidation reac-tions. Though they may not be involved in the
metabo-lism of a broad range or significant number of drugs, if they are the
primary enzyme responsible for the metab-olism of the drug of interest, then
their importance in that instance is obviously increased.
Regulation of the CYP Enzymes
CYP450 enzymes can be
regulated by the presence of other drugs or by disease states. This regulation
can ei-ther decrease or increase enzyme function, depending on the modulating
agent. These phenomena are com-monly referred to as enzyme inhibition and
enzyme in-duction, respectively.
Enzyme
Inhibition
Enzyme inhibition is the most
frequently observed re-sult of CYP modulation and is the primary mechanism for
drug–drug pharmacokinetic interactions. The most common type of inhibition is
simple competitive inhibi-tion, wherein two drugs are vying for the same active
site and the drug with the highest affinity for the site wins out. In this
scenario, addition of a second drug with greater affinity for the enzyme
inhibits metabolism of the primary drug, and an elevated primary drug blood or
tissue concentration is the result. In the simplest case, each drug has its own
unique degree of affinity for the CYP enzyme active site, and the degree of
inhibition depends on how avidly the secondary (or effector) drug binds to the
enzyme active site. For example, ketocona-zole and triazolam compete for
binding to the CYP3A4 active site and thus exhibit their own unique rate of
me-tabolism. However, when given concomitantly, the me-tabolism of triazolam by
the CYP3A4 enzyme (essen-tially the only enzyme that metabolizes triazolam) is
decreased to such a degree that the patient is exposed to 17 times as much of
parent triazolam as when keto-conazole is not present. Table 4.3 lists the
common CYP isoforms and representative inhibitory agents.

A second type of CYP enzyme inhibition is mecha-nism-based inactivation (or suicide inactivation). In this type of inhibition, the effector compound (i.e., the in hibitor) is itself
metabolized by the enzyme to form a reactive species that binds irreversibly to
the enzyme and prevents any further metabolism by the enzyme. This
mechanism-based inactivation lasts for the life of the enzyme molecule and thus
can be overcome only by the proteolytic degradation of that particular enzyme
molecule and subsequent synthesis of new enzyme pro-tein. A drug that is
commonly used in clinical practice and yet is known to be a mechanism-based
inactivator of CYP3A4 is the antibiotic erythromycin.
Enzyme
Induction
Induction of
drug-metabolizing activity can be due ei-ther to synthesis of new enzyme
protein or to a decrease in the proteolytic degradation of the enzyme.
Increased enzyme synthesis is the result of an increase in messen-ger RNA
(mRNA) production (transcription) or in the translation of mRNA into protein.
Regardless of the mechanism, the net result of enzyme induction is the
in-creased turnover (metabolism) of substrate. Whereas one frequently
associates enzyme inhibition with an in-crease in potential for toxicity,
enzyme induction is most commonly associated with therapeutic failure due to
in-ability to achieve required drug concentrations.
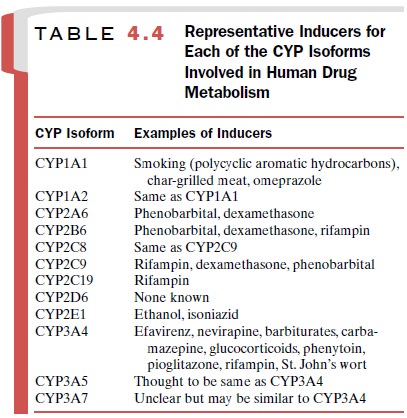
Table 4.4 lists
representative inducers of each of the CYP isoforms. No inducers of CYP2D6 have
been iden-tified.
The time course of enzyme
induction is important, since it may play a prominent role in the duration of
the effect and therefore the potential onset and offset of the drug
interaction. Both time required for synthesis of new enzyme protein (transcription
and translation) and the half-life of the inducing drug affect the time course
of induction. An enzyme with a slower turnover rate will require a longer time
before induction reaches equilibrium (steady state), and conversely, a faster
turnover rate will result in a more rapid induction. With respect to the drug
inducer, drugs with a shorter half-life will reach equilibrium concentrations
sooner (less time to steady state) and thus result in a more rapid maximal
induction, with the opposite being true for drugs with a longer half-life.
Flavin Monooxygenases
The flavin monooxygenases
(FMOs) are a family of five enzymes (FMO 1–5) that operate in a manner
analo-gous to the cytochrome P450 enzymes in that they oxi-dize the drug
compound in an effort to increase its elim-ination. Though they possess broad
substrate specificity, in general they do not play a major role in the
metabo-lism of drugs but appear to be more involved in the me-tabolism of
environmental chemicals and toxins.
Related Topics