Chapter: Essential Clinical Immunology: Immune-Mediated Neurological Syndromes
Myasthenia Gravis
MYASTHENIA GRAVIS
MG is an autoimmune disease that causes muscle
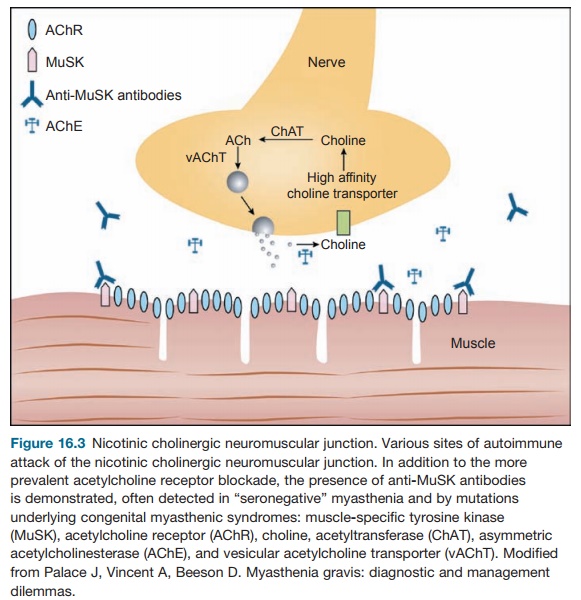
weakness believed to be primar-ily mediated by antibodies to the acetyl-choline
receptor (AChR) on the motor endplate of muscles (Figure 16.3). There are four
criteria that meet the definition of an antibody-mediated autoimmune disease
and MG meets all of them. First, the presumed antibody is present in 80 to 90
percent of patients. Second, the clinical syndrome can be passively transferred
to an animal model. Third, injection of the human antigen, in this case the
AChR into an animal can produce a model of the dis-ease. Fourth, an improvement
in the clinical disease state is associated with a decrease in antibody levels.
In the case of MG, a major diagnostic test after
physical exam would be to assay for the AChR antibody. The more severe the
clinical manifestation, the more likely the antibody will be detected and is
diag-nostic of the illness. However, absence of the antibody does not exclude
MG (see discussion about seronegative MG). Clinically, these patients present
with a muscle weakness only, associated with demonstrable fatigability of the
muscles on exam. For instance, if the extraocular muscles are involved,
weakness can be detected by the development of lid pto-sis or drooping when
having the patient sustain an upward gaze. Patients usually report a decrease
in strength later in the day, a hallmark symptom of this disease, rather than
uniform weakness through-out the day. Weakness often begins in the ocular
muscles producing double vision or a drooping lid but can go on to cause
weakness of the muscles of speech and swallowing (bulbar MG) as well as the
extremities. The respiratory muscles can be affected later on, in the setting
of a “myasthenic crisis,” which can be life threatening.
In infants, congenital myasthenia occurs from
transplacental passage of anti-bodies from a woman with MG or may be caused by
a mutation of the neuromuscu-lar junction itself. It is accepted as
conven-tional wisdom in the medical community that MG is antibody mediated
because an autoantibody specifically reactive to the acetylcholine receptor in
the neuromus-cular junction is present in 80–90 percent of patients. Reduction
of this antibody is associated with improvement in clinical symptoms. A
decrease in the number of acetylcholine receptors has been shown to occur
directly because of binding of anti-body to the receptors, possibly by
cluster-ing these receptors. These receptors may be destroyed ultimately via
the activation of the complement system.
The Lambert-Eaton syndrome is an immunological and
clinical variation of MG and is a paraneoplastic syndrome in the setting of
small cell lung cancer (70 percent of patients). More than 90 percent of
patients with this syndrome have autoanti-bodies to P/Q-type calcium channels.
Patients usually present with hip girdle weakness,
which is more notable in the morning and improves with exercise and over the
course of the day. Autonomic dysfunction is also found in addition to muscle
weakness.
Animal models for myasthenia gravis can be produced
by injecting the human antigen or by passively transferring the disease to an
animal by injection of the antibodies. However, the antibodies are far from
homogeneous. The subtypes of anti-bodies may vary among patients and even among
muscles within the same patient. The antibody may also have variations in light
chain type and subclass within one patient. Most likely these variations are
due to the receptor on the muscle, which consists of five subunits. These
subunits associate to form a transmembrane ion channel. It is likely that the
populations of B lymphocytes, producing the receptor antibodies, are also
heterogeneous.
T lymphocytes are also felt to play an important
role in MG, although they are not found in biopsy specimens. Their main
function in this setting might be to stimu-late the B cells. It has been
demonstrated that anti-T-cell antibodies may be used for immunoregulation in
this disease. The naturally occurring anti-T-cell receptor (anti-V beta 5.1)
IgG antibody is found to occur in those patients with milder disease and less
so in more severe MG. Although this antibody occurs in higher titers in MG
patients than in controls, the higher the antibody titer, the less severe are the
clinical symptoms, suggesting that therapy targeting pathogenic T-cell
receptors (TCRs) may be a useful avenue of exploration.
The thymus gland has been considered to be the
source of the autoimmune state of patients with MG. Most patients with MG have
thymic hyperplasia and 10 to 12 per-cent have a thymoma. The thymus gland
contains myoid-type cells, which have striations and acetylcholine receptors.
The thymic cells may act as antigen-present-ing cells with MHC class II
molecules. Cathepsin V, an enzyme responsible for cleaving the invariant chain
in the antigen-presenting cleft of the MCH II molecule is overexpressed in MG
patients in the thy-mic tissue and in the thymoma when pres-ent. However, mRNA
and cathepsin V pro-tein are not expressed in patients who have a thymoma but
do not have MG. Patients who have thymoma also may have anti-bodies directed
against muscle antigens as titin or the ryanodine receptor in addition to the
acetylcholine receptor.
The thymus gland may also play a role because of
the presence of acetylcho-line receptor on the myoid cells. Theories include
the possibility that a virus might alter the myoid cells, and the proximity to
the antigen presenting cells and helper T cells in the gland increases the
possibil-ity of an autoimmune response. Molecular mimicry may play a role as
well.
Herpes viruses and bacteria have been shown to
share cross-reactivity with the acetylcholine receptor. A genetic
pre-disposition is most likely a requisite of acquiring the disease. HLA types
have been associated with MG such as HLA-B8, DRw3, and DQw2. Other autoimmune
dis-eases are found to occur concomitantly in these patients or in their
families such as lupus, rheumatoid arthritis, and Graves’ disease.
There is also a subset of patients (10–20 percent)
with clinical MG, who do not produce antibodies to the acetyl-choline receptor.
They are designated as “antibody-negative myasthenia.” This may be a misnomer.
Some patients (40–70 percent) with this disorder produce anti-bodies to another
antigen, the MuSK. The antibodies are of the subclass IgG4 and strongly
activate complement cascade. In contrast, the ACh-R-Abs are primarily of the
IgG1 and IgG3 subclasses, which can also fix complement. The MuSK antibody is
also found in 10 percent of patients with antibody-positive MG, or alone
without the AChR-Ab.
Of interest, although no animal studies have shown
that this particular antibody can cause muscle weakness, a myasthenic syndrome
can be passively transferred with plasma from these patients into an animal
model. In addition, seronegative patients respond to plasmapheresis and
immunosuppressive therapy, similar to seropositive MG.
The experimental animal model of MG, experimental
autoimmune MG (EAMG) requires CD4+ helper T cells needed for the antibody-mediated
autoimmune response. In mice, the T-cell response to the AchR is predominantly
to a single peptide in about 50 percent of the cells. These T cells use a
restricted set of TCR genes and have a conserved CDR3 region.
This is in contrast to the human manifes-tation of
the disease, where AChR-specific T cells are very low in frequency. The
spe-cific T cells, when they have been cloned from patients are heterogeneous
in MHC restriction, and recognize various epitopes of the AChR.
There are three levels to treatment of myasthenic
patients, cholinesterase inhibitors, thymectomy, and immuno-suppression. The
mainstay of medica-tions that treat the symptoms, but not the course of the
disease, are the cholin-esterase inhibitors. These medications increase the
availability of acetylcholine in the neuromuscular junction and thereby help to
overcome the effect of decreased acetylcholine receptors due to the
autoan-tibodies, AChR-Ab.
However, while this medication may maintain
patients on a stable course for some time, they will not induce remis-sion of
the disease. The two approaches to this goal are thymectomy and
immuno-suppressants. The reason for thymectomy is that most MG patients have
thymic abnormalities, either hyperplasia (60–70 percent) or a thymoma (10–12
percent). Computerized tomography or MRI scan-ning of the mediastinum should be
routine in all patients suspected of MG. Although the clinical response is very
variable
according to the literature, thymectomy is the most
frequently used treatment for myasthenia and is generally suggested to be
performed very early. The rationale is that the thymus gland serves as a source
for the antigens involved in the autoim-mune response detected in patients,
even without the presence of thymoma.
The third arm of treatment is immuno-suppression.
Steroids improve 45 percent of patients and cause remission in 30 per-cent.
Steroids can initially cause a wors-ening of the clinical symptoms in the first
few weeks, which is unique to this dis-ease, and patients are often initiated
on them in a hospital setting. Plasmaphore-sis may be performed preventively
before steroids are initiated. Patients are often on steroids for one or two
years before tapering is begun. Immunosuppressive agents, such as azathioprine
or cyclospo-rine, are also administered in some cases because they act as
T-cell suppressors. The antibody production in this disease is T-cell
dependent, as demonstrated in the animal model.
Plasmapheresis or intravenous immu-noglobulin is
sometimes used in the set-ting of a clinical crisis. The plasmapho-resis
removes the AChR-antibody from the circulation and the clinical response
correlates with the decrease in antibody titer. Sometimes the plasma is
processed through a staph-protein A immunoabsor-bant column to more effectively
remove the IgG. Clinical trials are also under way using an AChR-Ab specific
immunoab-sorbant column. Intravenous immuno-globulin also has been demonstrated
to reverse an acute exacerbation for reasons unknown. For both treatments,
while the response time is rapid, a matter of days, the result is short lived,
lasting only a few months. It is hoped that within that time
Related Topics