Chapter: Essential Clinical Immunology: Immune-Mediated Neurological Syndromes
Guillain-Barré Syndrome
GUILLAIN-BARRÉ SYNDROME
GBS is a hallmark disease of molecular mimicry,
resulting in several different clinical manifestations. It was initially
characterized as a disease of the periph-eral nervous system because of the
earli-est detection of cases of flaccid paralysis in 1859 by Landry. By 1916,
Guillain, Barré, and Strohl had characterized the disease as having a rapidly
developing symmetri-cal flaccid paralysis, loss of reflexes, and autonomic
dysfunction, followed by a spontaneous remission. There is a pathog-nomonic
dissociation in the spinal fluid of the presence of high protein, without a
concomitant significantly elevated cell count. The category of pathology is
primar-ily a cellular immune system–mediated demyelinating disease with infiltration
of nerves by macrophages and lymphocytes (see Figure 16.1).
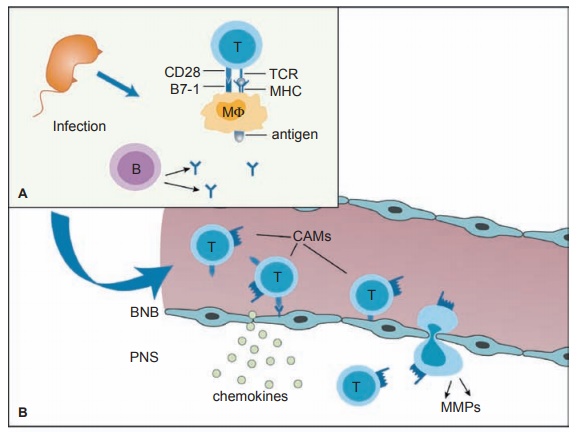
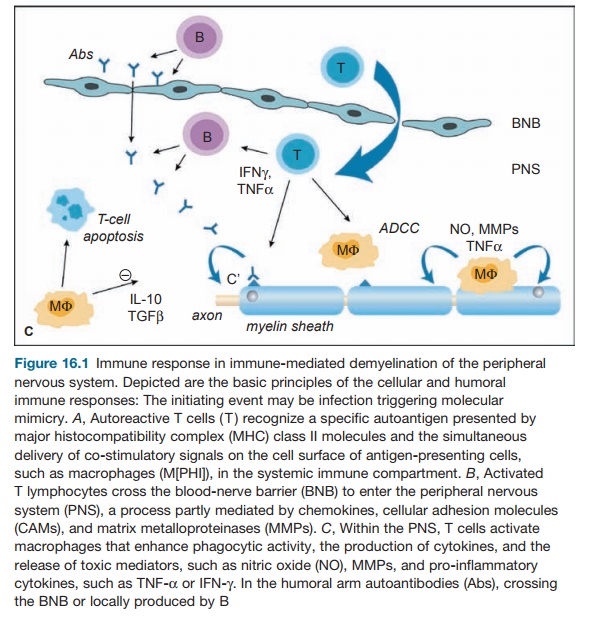
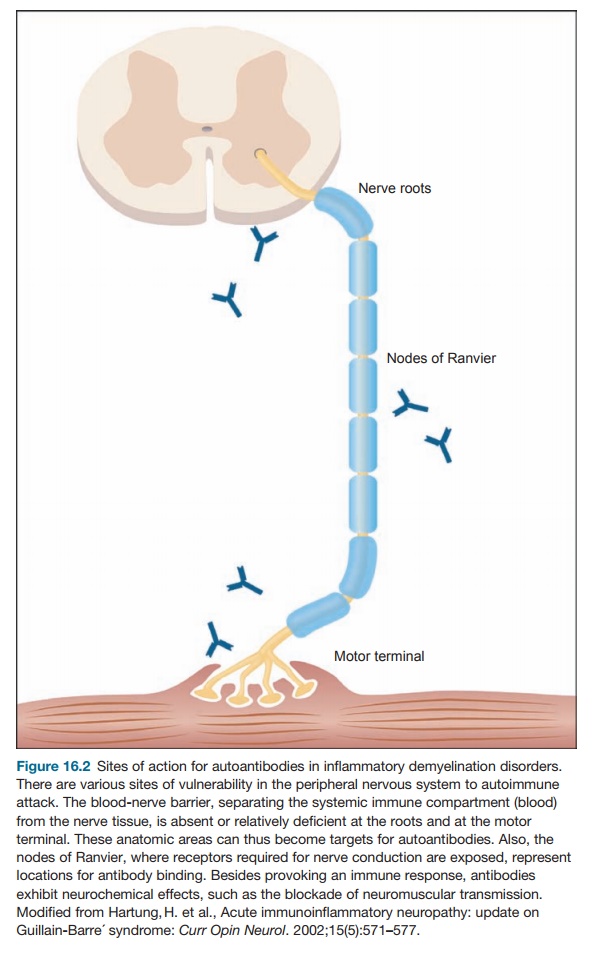
The humoral and cellular target is myelin directly
or the Schwann cells that produce myelin in the peripheral nervous system (see
Figure 16.2). The other name for this phenomenon is acute inflam-matory
demyelinating polyneuropathy (AIDP). Similar in pathology, but mani-festing
differently clinically is the Miller-Fisher variant of GBS, described as the
triad opthalmoparesis, ataxia, and absent reflexes, which may then encompass
the clinical AIDP form. Another variant of the disease includes the finding of
cranial nerve abnormalities, which can also evolve into AIDP. These variants
have variations in their immunopathology as well.
Most patients present with neurologi-cal complaints
two to four weeks after a mild gastrointestinal or upper respiratory illness.
The first symptoms may be tin-gling in the extremities, followed by weak-ness
in the legs and loss of reflexes, all of which may develop rapidly and ascend
into the arms, cranial nerves, and respira-tory muscles. The respiratory
decline can occur rapidly in some patients and may be life threatening. Twenty
to 30 percent require respiratory ventilators. In addition, patients can
develop life-threatening auto-nomic changes, including cardiac arrhyth-mias and
hypotension. Most patients are at their worst clinical state within two to four
weeks and start to recover within weeks to months. A chronic, or relapsing,
form of inflammatory demyelinating neuropathy also exists and is considered a
different disease (chronic inflammatory demyelinat-ing polyneuropathy).
Similar to MS, a demyelinating inflam-matory
disease of the CNS, an animal model exists for this peripheral demyelin-ating
disease entity, EAN. This is produced by inoculation with whole myelin or
spe-cific proteins of peripheral nervous system myelin in complete Freund’s
adjuvant. This produces a cell-mediated immune attack on native myelin
proteins.
However, more recently, basic prem-ises of this
disease have been redefined. It is now recognized that there may be a dis-tinct
axonal form of GBS. Electromyogra-phy has been able to distinguish these two
forms. An attack against myelin and axons may be found concurrently in the same
patient, with both demyelination and peri-axonal macrophages.
Molecular mimicry has been found to be the major
pathogenic force and the basis for autoimmunity. Various preced-ing infections
can initiate the immune response that causes the clinical state of GBS. The two
major agents are Campylo-bacter and Cytomegalovirus, with Epstein-Barr
virus considered an inciting agent as well. Specific features of the incit-ing
organism may play a major role in the development of GBS. For instance,
although Campylobacter jejuni often
may cause diarrhea, the strains that have been linked to GBS (O-serotypes) are
different from those strains causing diarrhea only but are genetically similar
to each other. Only 70 percent of those infected with Campylobacter in the presence of GBS give a previous history of gastrointestinal com-plaints in the three
months before the neu-rological onset, but the risk of developing GBS within
two months of a symptomatic Campylobacter
infection is 100-fold higher than
the general population. GBS follow-ing C.
jejuni and intestinal symptoms are felt to have a more dire clinical
course, involving axonal destruction in addition to demyelination.
The Miller-Fisher variant, which involves primarily
the cranial nerves, dem-onstrates the close association between antiganglioside
antibodies and a specific syndrome. Anti-GQ1b has a high specificity and
sensitivity for this symptom complex. GQ1b is a ganglioside that is a component
of cranial nerve myelin. Similarly, anti-GM1 (anti-GD1a) antibodies have been
linked specifically to the axonal form of the disease, acute motor axonal
neuropathy. Although GM1 antigens can be found on motor and sensory nerves,
there are vari-able reports in the literature about whether these antigens are
on the axon or in the myelin sheath. However, antibodies to GM1 can bind to the
nodes of Ranvier and activate complement. Other factors may be necessary for
the disease to occur, as many patients with Campylobacter
infections pro-duce antibodies to GM1 ganglioside but do not develop
neurological symptoms. Because the disease can improve rapidly, an association
between sodium channels and the antibody has been studied, but there has been
no consistent association with sodium channels.
Influenza vaccine has been implicated as a
causative agent for GBS as well. How-ever, careful epidemiological studies have
not borne this out. Combining the 1992–1993
The host may play a significant role in this
disease as well. GBS has been associ-ated with certain major histocompatibility
complex (MHC) class II genes and may determine a heightened immune response to C. jejuni, resulting in GBS in these
patients compared with controls who do not go on to develop the neurological
disease after exposure to Campylobacter.
GBS patients may be high producers of tumor necrosis factor alpha 2 compared
with controls.
In the Fisher variant, anti-GQ1b anti-bodies affect
acetylcholine release at the nerve terminals. In one mouse model of disease,
applying serum from patients on a mouse model is similar to that of the black
widow spider venom – the inception of a large release of acetylcholine, which
is cal-cium dependent. Complement is required for this reaction, either by the
classical or alternative pathway. Presumably, humoral immunity is the inciting
factor.
For the classic AIDP form of the dis-ease, the
target antigen is still unknown. Two candidate antigens are the myelin protein
PMP22 and the heparin sulfate glycosoaminoglycans. The virulence of some C. jejuni strains may be due to sev-eral
factors. First, specific cross-reactive antigens may be more common in these
strains. These strains may also have higher immunogenicity or may have greater
inva-siveness. The O-19 strains (overly pre-sented in GBS) do have GM1-like
epitopes composed of lipopolysaccharides. How-ever, other organisms do as well,
including those causing enteritis only, and humans are frequently exposed to C. jejuni with GM1 cross-reactivity.
Therefore, these epi-topes do not alone explain GBS.
Therapy for GBS has been directed at the immune
mechanisms, but interestingly, corticosteroids have been shown to have no
clinical effect, as has been established in many other neurological
manifestations of autoimmune mechanisms. Instead, the two mainstays of therapy
have been plasma exchange and intravenous immunoglobu-lins (IVIG). These
treatments are suggested for those with illness severe enough to limit walking
ability. Plasma exchange using fresh frozen plasma or albumin has been shown to
increase muscle strength and decrease the time needed for a respirator to
assist ventilation. Intravenous immune globulin has similar efficacy, but there
is no additive effect of combining the two therapies. In animal models, IVIG
has been shown to neutralize neuromuscular block-ing antibodies and inhibit
binding of anti-GQ1b antibodies to GQ1b. These prevent initiation of the
destructive complement cascade. Overall, 80 percent of patients recover fully
or are left with only minor deficits. Up to 10 percent are left with weakness,
imbalance, or loss of sensation and 3 to 8 percent die from sepsis, pulmo-nary
emboli, cardiac arrest, or acute respi-ratory distress syndrome even despite
care in an intensive care unit. These patients are usually older, have
preexisting pulmonary disease, or were on prolonged respirator assistance.
Related Topics