Chapter: Medical Microbiology: An Introduction to Infectious Diseases: Neisseria
Meningo Coccal Disease
MENINGO COCCAL DISEASE
Meningococci are usually quiescent members of the nasopharyngeal flora but may produce fulminant infection of the bloodstream and/or central nervous system CNS). There is little warning; localized infections that precede systemic spread are rarely recognized. The major disease is an acute, purulent meningitis with fever, headache, seizures, and mental signs secondary to inflammation and in- creased intracranial pressure. Even when the CNS is not involved, N. meningitides infections have a marked tendency to be accompanied by rash, purpura, thrombo- cytopenia, and other manifestations associated with endotoxemia. This bacterium causes one of the few infections in which patients may progress from normal health to death in less than a day.
EPIDEMIOLOGY
Meningococci are found in the nasopharyngeal flora of approximately 10% of healthy in-dividuals. Transmission occurs by inhalation of aerosolized respiratory droplets. Close, prolonged contact such as occurs in families and closed populations promotes transmis-sion. The estimated attack rate among family members residing with an index case is 1000 times higher than in the general population; this fact is evidence of the contagious nature of meningococcal infection. Other factors that foster transmission are contact with a virulent strain and susceptibility (lack of protective antibody). Typical settings of larger outbreaks are schools, dormitories, and camps for military recruits. In these close living circumstances, N. meningitidis spreads readily among newly exposed individuals, but disease develops only in those who lack group-specific antibody.
The annual incidence of meningococcal infections in the United States varies be-tween 0.5 and 1.5 cases per 100,000 population. Most cases are in children under 6 years of age. They occur as isolated cases, as sporadic small epidemics, or in small family or closed-population (school or day-care center) outbreaks. B, C, and Y are the most common serogroups involved. Group A strains are generally rare but historically have a more ominous epidemiologic potential. For unknown reasons, group A meningo-cocci have the capability to cause widespread epidemics sweeping through communities, even countries. In the past these have appeared in 8- to 12-year cycles. It has been more than 40 years since group A strains have been responsible for significant disease in the United States, although epidemics have occurred in Brazil, China, the Sudan, Kenya, and South Africa.
PATHOGENESIS
The meningococcus is an exclusively human parasite; it can either exist as an apparently harmless member of the normal flora or produce acute disease. For most individuals, the carrier state is associated with acquisition of protective antibodies, but for some, spread from the nasopharynx to produce bacteremia, endotoxemia, and meningitis takes place too quickly for immunity to develop. Meningococci use pili for initial attachment to the mi-crovilli of the nonciliated nasopharyngeal epithelium as a prelude to invasion. In the inva-sion process, the microvilli come in close contact with the bacteria, which then enter the cells in membrane-bound vesicles. Once inside meningococci quickly pass through the cy-toplasm, exiting into the submucosa on the other side. In the process they damage the cili-ated cells, possibly by direct release of endotoxin.
Once meningococci gain access to the submucosa, their ability to produce disease is enhanced by several factors that allow them to scavenge essential nutrients and evade the host immune response. One critical nutrient, iron, is supplied by N. meningitidis proteins, which are able to acquire it from the human iron transport protein transferrin. As with other encapsulated bacteria, the polysaccharide capsule enables meningococci to resist complement-mediated bactericidal activity and subsequent neutrophil phagocytosis. Meningococcal (and gonococcal) LPS/LOS also has features that facilitate evasion of host immune responses. Its chemical structure mimics sphingolipids found in the human brain enough for them to be recognized as self by the immune system. In addition, meningococci are able to incorporate sialic acid from host substrates as terminal substitu-tions of their LOS side chains. This sialyated LOS is able to downregulate complement deposition by binding serum factor H in a manner already described for streptococcal sur-face molecules such as group B streptococcal capsular sialic acid . The capsules of group B and C meningococci are also polymers of sialic acid.
The most serious manifestations of meningococcal disease are related to its spread to the bloodstream and, its namesake, the meninges. The exact mechanism of CNS invasion is unclear but is probably related to the level of the bacteremia. It occurs in the choroid plexus with its exceptionally high rate of blood flow. After CNS invasion, an intense sub-arachnoid space inflammatory response is generated, induced by the release of cell wall peptidoglycan fragments, LPS, and possibly other virulence factors causing the release of inflammatory cytokines. A prominent feature of meningococcal disease with or without CNS invasion is disseminated, potent, endotoxic activity (see Manifestations). When grown in culture, N. meningitidis readily releases endotoxin-containing blebs of its outer membrane from the cell surface as shown in Fig 20 – 1. It is not known whether this oc-curs in vivo, but the model of the meningococcus as a hyperproducer of LPS endotoxin certainly fits with its most serious disease manifestations.
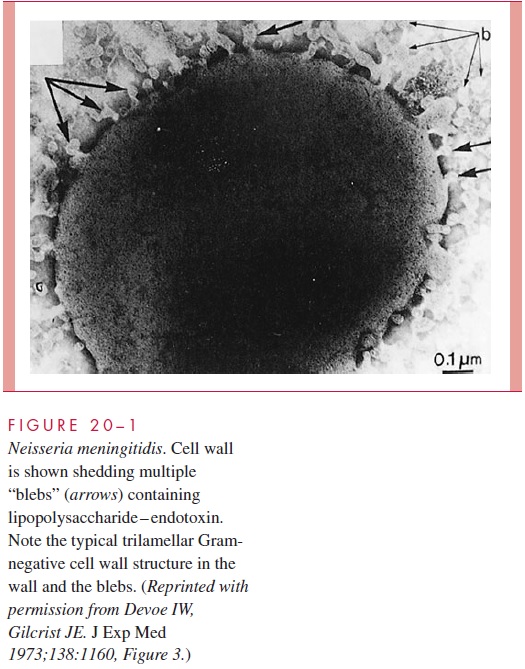
IMMUNITY
Immunity to meningococcal infections is related to group-specific antipolysaccharide an-tibody, which is bactericidal and facilitates phagocytosis. The bactericidal activity is due to complement-mediated cell lysis via the classical complement pathway. Individuals with deficiencies in the terminal complement components have an enhanced risk for meningococcal disease but not for other polysaccharide capsule pathogens such asHaemophilus influenzae type b .
The peak incidence of serious infection is between 6 months and 2 years of age. This corresponds to the nadir in the prevalence of antibody in the general population, which is the time between loss of transplacental antibody and the appearance of naturally acquired antibody (Fig 20 – 2). By adult life, serum antibody to one or more meningococcal serogroups is usually present but an immune deficit to the other serogroups remains. In-fections appear when populations carrying virulent strains mix with susceptible individu-als lacking group-specific antibody.
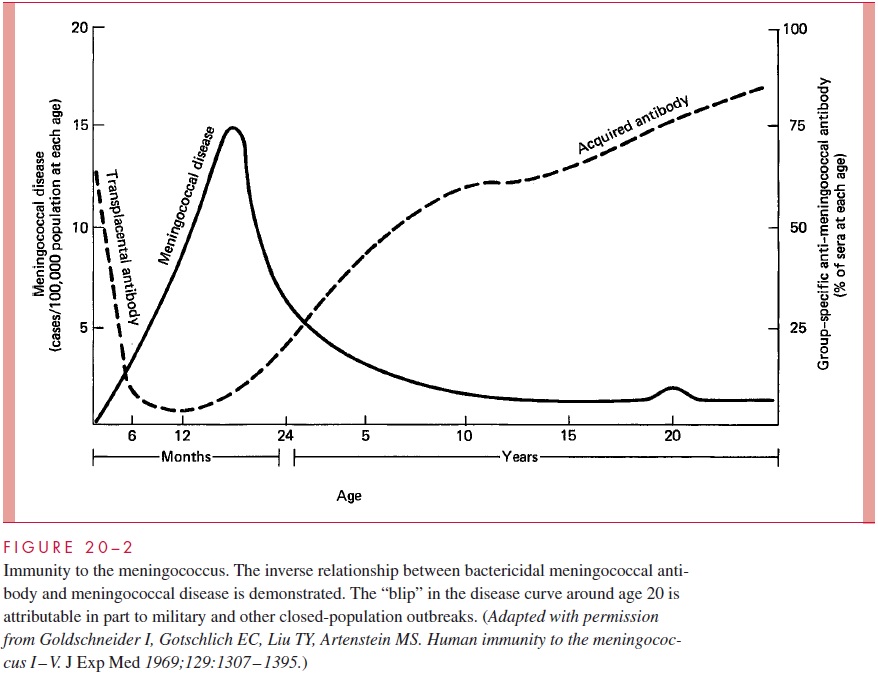
Protective antibody is stimulated by infection and through the carrier state, which pro-duces immunity within a few weeks. Natural immunization may not require colonization with every serogroup or even with N. meningitidis, because antibody may be produced in response to cross-reactive polysaccharides possessed by otherNeisseria or even other genera. For example, Escherichia coli strains of a particular serotype (K1) have a poly-saccharide capsule identical to that of the group B meningococcus. These E. coli also have enhanced potential to produce meningitis in neonates.
Purified capsular polysaccharides are immunogenic, generating T cell – independent immune responses in which IgG2 is the predominant antibody. As with other polysaccha-ride immunogens, these responses are not strong, particularly in early childhood when there is a relative deficiency of IgG2. The group B polysaccharide differs from that of the other groups in failing to stimulate bactericidal antibody at all. This is believed to be due to the similarity of its sialic acid polymer to human brain antigens. That is, like the sialated LOS, it may be recognized as self by the immune system.
Exposed outer membrane proteins have also been shown to stimulate bactericidal an-tibody. Antibody directed against the porin PorA has demonstrated protection in animal models. PorA is present in the outer membrane of almost all meningococcal isolates but is subject to considerable antigenic variation.
Related Topics