Chapter: Medical Microbiology: An Introduction to Infectious Diseases: Antimicrobial Resistance
Mechanisms of Resistance - Bacterial Resistance to Antimicrobics
BACTERIAL RESISTANCE TO ANTIMICROBICS
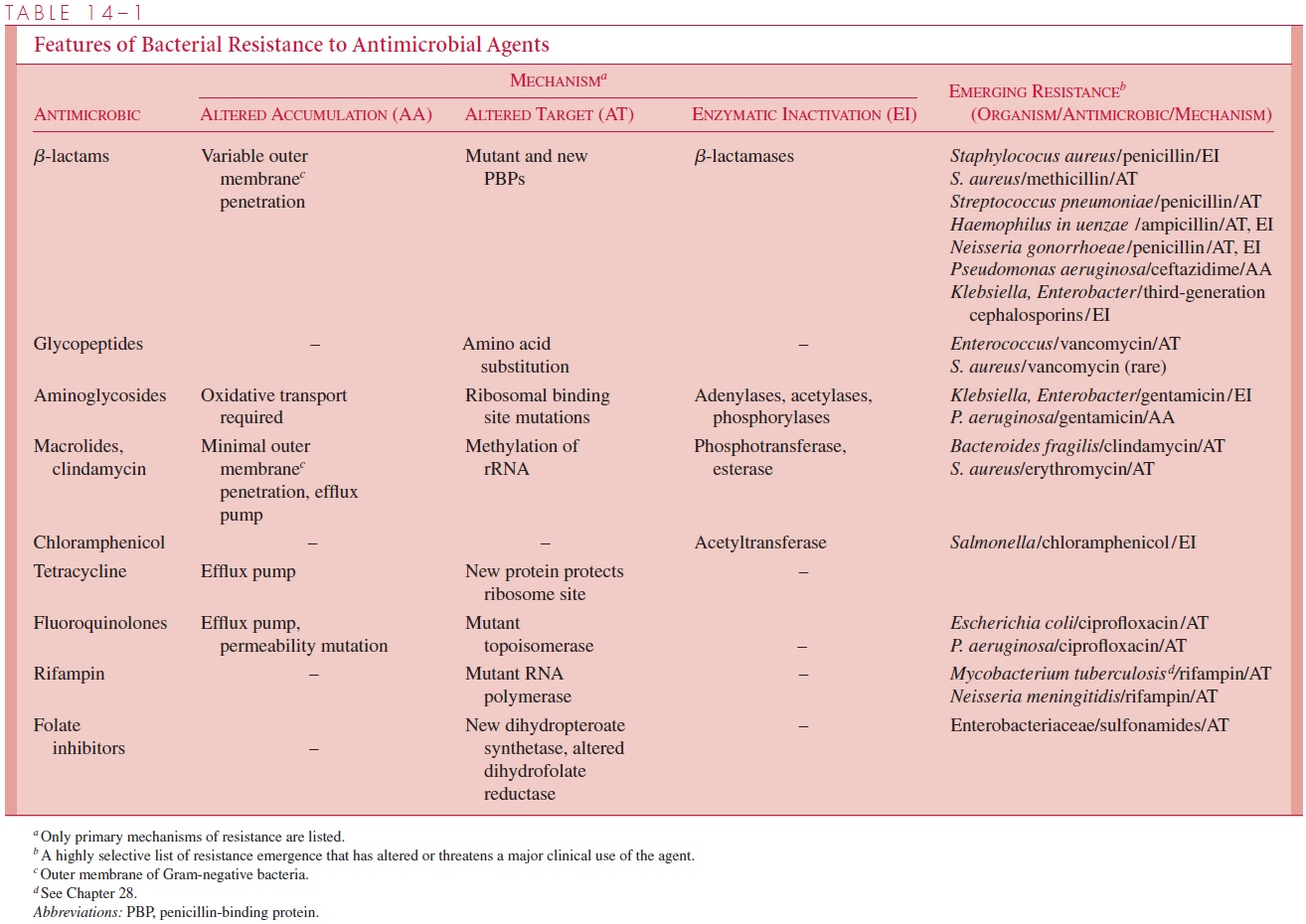
The seemingly perfect nature of antimicrobics, originally hailed as “wonder drugs,” has been steadily eroded by the appearance of strains resistant to their action. This resistance may be inherent to the organism or appear in a previously susceptible species by mutation or the acquisition of new genes. The mechanisms by which bacteria develop resistance and how this resistance is spread are of great interest for the continued use of current agents and to develop strategies for the development of new antimicrobics. The following sections discuss the biochemical mechanisms of resistance, how resistance is genetically controlled, and how resistant strains survive and spread in our society. How these features relate to the antimicrobic groups is summarized in Table 14 – 1on specific bacteria.
Antimicrobial resistance has survival value for the organism, and its expression in the medical setting requires that virulence be retained despite the change that mediates resis-tance. There are no direct connections between resistance and virulence. Resistant bacte-ria may have increased opportunities to produce disease, but the disease is the same as that produced by the susceptible bacteria’s counterpart.
Mechanisms of Resistance
The major mechanisms of bacterial resistance are (1) accumulation barriers to an antimi-crobic due to impermeability or active efflux; (2) alterations of an antimicrobic target, which render it insusceptible; and (3) inactivation of an antimicrobic by an enzyme pro-duced by the microorganism. Changes in metabolic pathways can also translate into resis-tance in a few antimicrobic – organism combinations.
Accumulation Barriers (Fig 14 – 3)
An effective antimicrobic must enter the bacterial cell and achieve concentrations suffi-cient to act on its target. The cell wall, particularly the outer membrane, of Gram-negative bacteria presents a formidable barrier for access to the interior of the cell. Outer mem-brane protein porin channels may allow penetration depending on the size, charge, degree of hydrophobicity, or general molecular configuration of the molecule. This is a major reason for inherent resistance to antimicrobics, but these transport characteristics may change even in typically susceptible species due to mutations in the porin proteins.
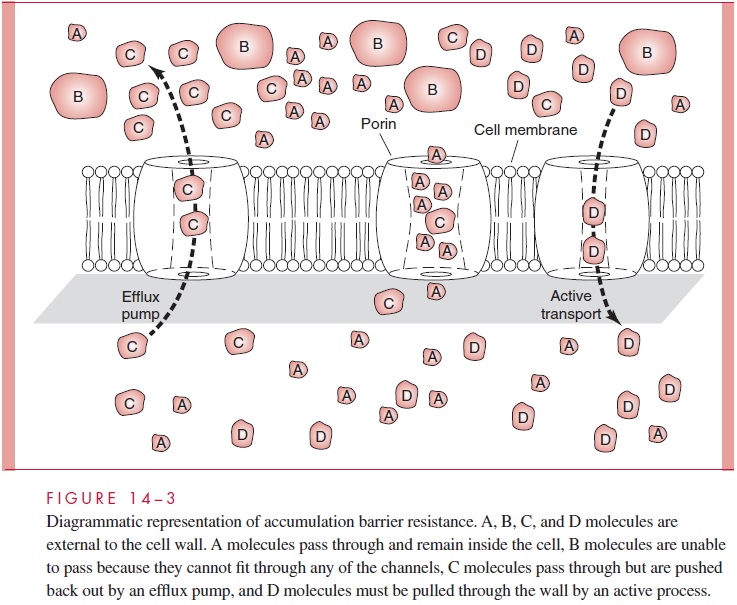
For example, strains of Pseudomonas aeruginosa commonly develop resistance to imipenem due to loss of the outer membrane protein most important in its penetration.
As mentioned above, some antimicrobics must be actively transported into the cell. Bacteria such as streptococci, enterococci, and anaerobes, which lack the necessary ox-idative pathways for transport of aminoglycosides, are resistant. Conversely, some antimi-crobics are actively transported out of the cell. A number of bacterial species have energy-dependent efflux mechanisms that pump either tetracyclines or fluoroquinolones from the cell.
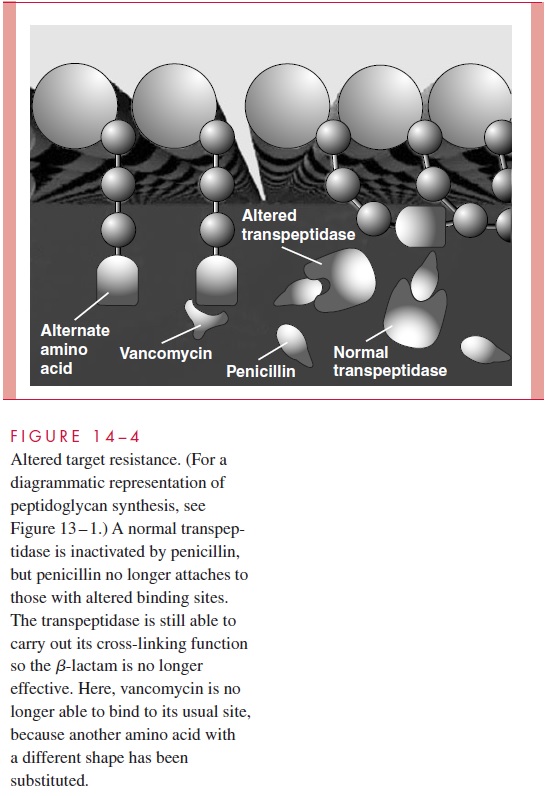
Altered Target (Fig 14–4)
Once in the cell, antimicrobics act by binding and inactivating their target, which is typically a crucial enzyme or ribosomal site. If the target is altered in a way that decreases its affinity for the antimicrobic, the inhibitory effect will be proportionately decreased. Substitution of a single amino acid at a certain location in a protein can alter its binding to the antimicrobic without affecting its function in the bacterial cell.
If an alteration at a single site of the target does not render it susceptible to the antimicrobic, mutation to resistance can occur in a single step, even during therapy. This occurred with the early aminoglycosides (streptomycin), which bound to a single riboso-mal site, and the first quinolone (nalidixic acid), which attached to only one of the four topoisomerase subunits. Newer agents in each class bind at multiple sites on their target, making mutation to resistance statistically improbable.
One of the most important examples of altered target involves the bβ-lactam family and the peptidoglycan transpeptidase penicillin-binding proteins (PBPs) on which they act. In widely divergent Gram-positive and Gram-negative species, changes in one or more of these proteins have been correlated with decreased susceptibility to multiple b-lactams. These alterations were initially detected as changes in electrophoretic migra-tion of one or more PBPs using radiolabeled penicillin (hence the origin of the term PBP). These changes have now been traced to point mutations, substitutions of amino acid sequences, and even synthesis of a new enzyme.
Because the altered binding is not absolute, decreases in susceptibility are incremental and often small. Wild-type pneumococci and gonococci are inhibited by 0.06 μg/mL of penicillin, while those with altered PBPs have MICs of 0.1 to 8.0 μg/mL. At the lower end, these MICs still appear to be within therapeutic range but are associated with treat-ment failures, even when dosage is increased. Altered PBPs generally affect all b-lac-tams. Although the exact MICs vary, a strain with a 10-fold decrease in susceptibility to penicillin has decreased susceptibility to cephalosporins to about the same degree.
PBP alterations are the prime reason for emergence of penicillin-resistant pneumo-cocci and methicillin-resistant Staphylococcus aureus (MRSA). They are one of multiple mechanisms of resistance for a variety of other bacteria including enterococci, gono-cocci, Haemophilus in uenzae, and many other Gram-positive and Gram-negative species.
Alteration of the target does not require mutation and can occur by the action of a new enzyme produced by the bacteria. Vancomycin-resistant enterococci have enzyme sys-tems that substitute an amino acid in the terminal position of the peptidoglycan side chain (alanyl lysine for alanyl alanine). Vancomycin does not bind to the alternate amino acid, and these strains are resistant. Resistance to sulfonamides and trimethoprim occurs by ac-quisition of new enzymes with low affinity for these agents but still allows bacterial cells to carry out their respective functions in the folate synthesis pathway.
Clindamycin resistance involves an enzyme that methylates ribosomal RNA, prevent-ing attachment. This modification also confers resistance to erythromycin and other macrolides, because they share binding sites. Interestingly, induction with erythromycin leads to clindamycin resistance, although the reverse is unusual.
Enzymatic Inactivation (Fig 14–5)
Enzymatic inactivation of the invading antimicrobic is the most powerful and robust of the resistance mechanisms. Literally hundreds of distinct enzymes produced by resistant bacteria may inactivate the antimicrobic in the cell, in the periplasmic space, or outside the cell. They may act on the antimicrobic molecule by disrupting its structure or by cat-alyzing a reaction that chemically modifies it.
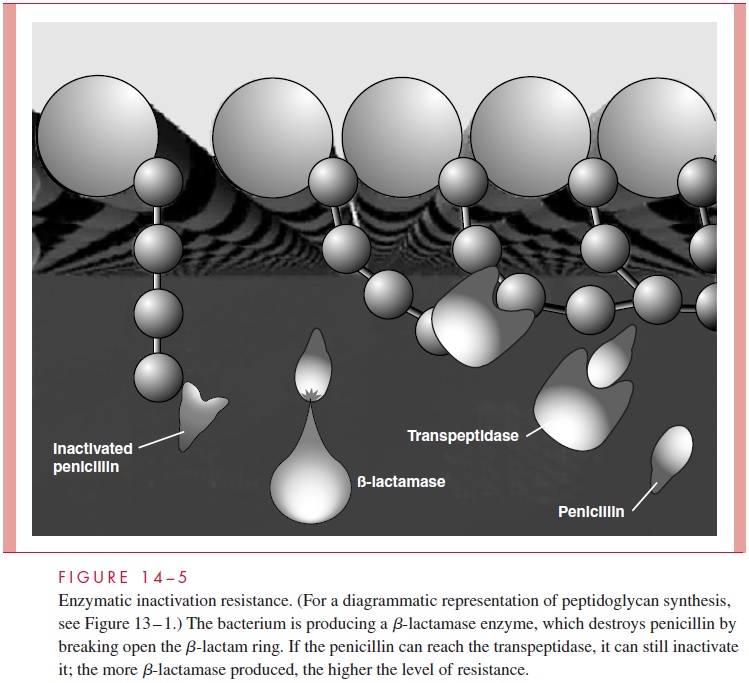
β-lactamases β-lactamase is a general term referring to any one of hundreds of bac-terial enzymes able to break open the β-lactam ring and inactivate various members of the β-lactamgroup. The first was discovered when penicillin-resistant strains of S. aureus emerged and were found to inactivate penicillin in vitro. The enzyme wascalled penicillinase, but with expansion of the β-lactam family and concomitant resis-tance, it has become clear that the situation is quite complex. Each β-lactamase is a dis-tinct enzyme with its own physical characteristics and substrate profile. For example, the original staphylococcal penicillinase is also active against ampicillin but not against me-thicillin or any cephalosporin. β-lactamases produced by Escherichia coli may add cephalosporinase activity but vary in their potency against individual first-, second-, third-, and fourth-generation cephalosporins. Some β-lactamases are bound by clavu-lanic acid, and others are not.
To keep track of the β-lactamase identifiers (eg, TEM-1, TEM-2, OXA, SVH), clas-sification schemes have been created based on molecular structure, substrate profile, and inducibility (ie, whether enzymes are inducible or produced constitutively). A con-sideration of β-lactamase classification is beyond the scope of this book, but some dis-cussion of the major types is useful. Gram-positive β-lactamases are exoenzymes with little activity against cephalosporins or the antistaphylococcal penicillins (methicillin, oxacillin). They are bound by β-lactamase inhibitors such as clavulanic acid. Gram-negative enzymes act in the periplasmic space and may have penicillinase and/or cephalosporinase activity. They may or may not be inhibited by clavulanic acid. Many of the Gram-negative β-lactamases are constitutively produced at very low levels but can be induced to high levels by exposure to a β-lactam agent. A newer class, called ex-tended-spectrum β-lactamases (ESBLs) because their range includes multiple cephalosporins, is particularly worrisome. The laboratory detection of ESBLs is com-plex because they are inducible enzymes, and the conditions for induction may not be met in the susceptibility test.
Bacteria that produce β-lactamases typically demonstrate high-level resistance with MICs far outside the therapeutic range. Even weak β-lactamase producers are considered resistant because the outcome of susceptibility tests (and presumably infected sites) is strongly influenced by the number of bacteria present. Rapid direct tests for β-lactamase can provide this information in a few minutes.
Modifying Enzymes The most common cause of acquired bacterial resistance to amino-glycosides is through production of one or more of over 50 enzymes that acetylate, adenylate, or phosphorylate hydroxyl or amino groups on the aminoglycoside molecule. The modifications take place in the cytosol or in close association with the cytoplasmic membrane. The resistance conveyed by these actions is usually high level; the chemically modified aminoglycoside no longer binds to the ribosome. As with the β-lactamases, the aminoglycoside-modifying enzymes represent a large and diverse group of bacterial pro-teins, each with its characteristic properties and substrate profile. Inactivating enzymes have been described for a number of other antimicrobics. Most of these act by chemically modifying the antimicrobic molecule in a manner similar to the aminoglycoside-modify-ing enzymes. The most clinically significant enzymes convey resistance to erythromycin (esterase, phosphotransferase) and chloramphenicol (acetyltransferase).
Related Topics