Chapter: Essential Clinical Immunology: Immunological Aspects of Renal Disease
Mechanisms of Immune Injury to the Kidneys
MECHANISMS OF IMMUNE INJURY TO THE
KIDNEYS
Cells of both innate and adaptive immu-nity mediate
human defense against microbes. The principal components of innate immunity are
composed of physical and chemical barriers (e.g., epithelial cells and
antimicrobial substances produced by the cells), phagocytic cells (neutrophils
and macrophages), natural killer cells, comple-ment systems, and cytokines.
These repre-sent the first line of defense against micro-bial agents. In
contrast, adaptive immunity is represented by B and T lymphocytes. These cells
are initially stimulated by expo-sure to offending agents. Subsequent expo-sure
to similar agents cause specific cells to mount a defense in increasing
magnitude.
Adaptive immunity is distinguished as having the
ability to “remember” specific molecules and therefore provide specific
immunity. Both types of immunity work in tandem to provide comprehensive
defense against offending microbes.
Diseases caused by immune responses are often
called hypersensitivity diseases.
These are classified according to the type of immune responses and the effector
mecha-nisms that are responsible for cell and tis-sue injury.
Immediate hypersensitivity or type I disease is
mediated by immunoglobulin E (IgE), which is produced in response to an
allergen. After exposure to a spe-cific allergen, TH2 cells
specific for the allergen are activated. IgE antibodies are then produced,
which subsequently bind to the Fc receptor of mast cells and basophils. This
binding releases biogenic amines (histamine), neutral serine prote-ases, lipid
mediators, and cytokines. Bio-genic amines and lipid mediators cause vascular
leakage, vasodilation, and air-way bronchoconstriction. Serine prote-ases cause
tissue damage. Cytokines are implicated in late phase reaction. Certain drugs,
especially methicillin, nonsteroi-dal agents, rifampin, sulfa, cimetidine,
cephalosporins, and 5-aminosalicylates, can cause allergic interstitial
nephritis. Affected patients present with acute renal failure three to five
days after intake of the offending drug. Fever, rash, hematu-ria, proteinuria,
and eosinophiluria are typical findings. On kidney biopsy, tubu-lointerstitial
inflammation is prominent with occasional demonstration of eosino-phils. The
glomeruli appear spared of any inflammatory effects. Treatment consists of
withholding the offending drug and use of steroids.
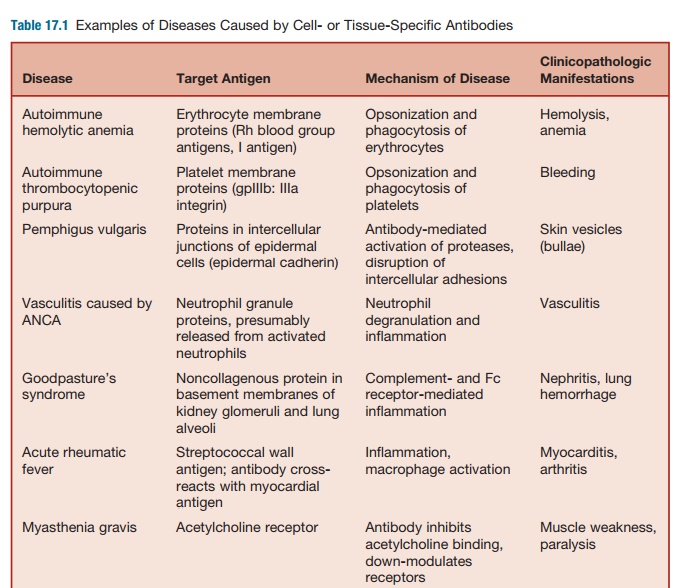
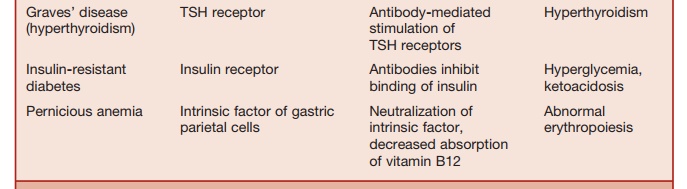
Antibody-mediated or type II disease is caused by
antibodies against fixed cell and tissue antigens. Although most cases
demonstrate the presence of autoantibod-ies, some antibodies can be produced by
a foreign antigen that is immunologically cross-reactive to human tissue. Three
mechanisms have been described to explain this phenomenon. First, antibodies
may opsonize cells or activate the complement system that eventually produces
comple-ment proteins that assist in opsonization of cells. Macrophages bind to
Fc receptors of antibodies or complement protein receptors to cause endocytosis
and destruction of the offending antigen. This appears to be the main mechanism
in hemolytic anemia and autoimmune thrombocytopenic purpura. Second, antibodies
bound in target tissues recruit neutrophils and macrophages by binding to Fc
receptors or by activating complement. Activated neutrophils and macrophages
release intracellular enzymes and reactive oxygen radicals that cause tissue
injury. Examples of glomerular dis-ease that can be explained by this pathway
include Goodpasture’s syndrome and anti-neutrophil cytoplasmic antibody (ANCA)
mediated disease. Third, antibodies may bind to normal cellular receptors and
inter-fere with their function to cause disease. However, no actual tissue
injury is dem-onstrated. Graves’ disease represents this mechanism where the
TSH receptor is tar-geted by the anti-TSH-receptor antibody. This mechanism
causes hyperthyroidism. No glomerular disease has been associated with this
mechanism (see Table 17.1).
Immune-complex-mediated, or type III, disease is
caused by deposition of antibod-ies bound to self- or foreign antigens into target
tissues. Although the glomerulus is a common target for immune complexes, other
organ systems are involved, which suggests the systemic nature of this
dis-ease. A classic example is the serum dis-ease or serum sickness, which was
origi-nally described by Clemens von Pirquet in 1911. During his time,
diphtheria infec-tions were treated with passive immuniza-tion using serum from
horses immunized with diphtheria toxin. He noted that joint inflammation
(arthritis), rash, and fever occurred in patients injected with the
anti-toxin-containing horse serum. On further investigation, similar symptoms
were seen in patients injected with horse serum with-out the antitoxin. The
symptoms usually occurred at least one week after the first injection and more
rapidly on subsequent injections. He concluded that the host had developed
antibodies to the horse serum, and deposition of antibody–serum protein
complexes (immune complexes) to differ-ent tissues of the host caused the
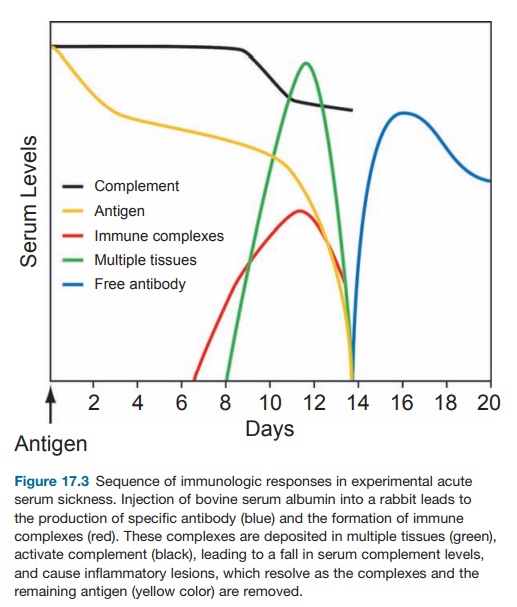
symp-toms described earlier. In experimental animals like the rabbit, injection
of a large dose of a foreign protein antigen leads to the formation of
antibodies against the antigen (see Figure 17.3). Subsequent for-mation of
antibody–antigen complexes leads to enhanced phagocytosis and clear-ance of the
antigen by the macrophages in the liver and the kidney. With subse-quent
injection of the antigen, more of the immune complexes are formed and may be
deposited in the vascular bed, renal glomeruli, and synovia. These activate the
complement, which leads to recruitment of inflammatory cells (predominantly
neutrophils) to cause injury to the affected tissues. Clinical and pathological
manifes-tations are vasculitis, glomerulonephri-tis, and arthritis. Clinical
symptoms are
T-cell-mediated, or type IV, hyper-sensitivity
diseases involve activation of CD4+ T cells of the TH1
subset and CD8+ T cells. Both types of T cells release interferon-γ and activate macrophages, which
can release tumor necrosis factor (TNF), interleukin-I (IL-1), and other
chemokines that are involved the inflammatory processes. In delayed-type
hypersensitivity (DTH), tissue injury is mediated by hydrolytic enzymes,
reactive oxygen intermediates, and nitric oxide.
There is
also up-regulation of adhesion molecules and class II molecules in vascular
endothelial cells. Chronic DTH processes cause fibrosis because of continuous
secretion of cytokines and growth factors. In some instances, CD8+
cytolytic T cells (CTL) directly kill target cells bearing class I MHC cells.
T-cell-mediated
renal diseases are best exemplified in kidney allograft rejection. In acute
allograft rejection, T cells react to alloantigens, including MHC molecules,
which reside on the vascular endothelial and renal parenchymal cells. Microvas-cular
endothelitis is an early finding in acute rejection. Later involvement of the
medium-sized arteries signifies severe rejection. Experimental evidence that points
to the involvement of CD8+ CTL in allograft rejection include the
presence of RNAs encoding CTL-specific genes (e.g., perforin and granzyme B),
presence of cel-lular infiltrates enriched with CTL popu-lation, and ability to
adoptively transfer alloreactive CD8+ CTL cells. CD4+ T
cells mediate rejection by secreting cytokines and inducing DTH-like reactions
in the allograft. Adoptive transfer of alloreactive CD4+ T cells in
experimental animals has been known to cause rejection of the allo-graft as
well. In chronic allograft rejection, histopathological findings are compatible
with by-products of a chronic inflamma-tory state (interstitial fibrosis,
vascular occlusion, and glomerulosclerosis) (Abbas and Lichtman 2005).
Related Topics