Chapter: Clinical Anesthesiology: Perioperative & Critical Care Medicine: Thermoregulation, Hypothermia, & Malignant Hyperthermia
Malignant Hyperthermia
MALIGNANT HYPERTHERMIA
Malignant hyperthermia (MH) is a rare (1:15,000 in pediatric patients
and 1:40,000 adult patients) genetic hypermetabolic muscle disease, the
charac-teristic phenotypical signs and symptoms of which most commonly appear
with exposure to inhaled general anesthetics or succinylcholine (triggering
agents). MH may occasionally present more than an hour after emergence from an
anesthetic, and rarely may occur without exposure to known trig-gering agents.
Most cases have been reported in young males;
almost none have been reported in infants, and few have been reported in the
elderly. Nevertheless, all ages and both sexes may be affected. The incidence
of MH varies greatly from country to country and even among different
geographic locali-ties within the same country, reflecting varying gene pools.
The upper Midwest appears to have the great-est incidence of MH in the United
States.
Pathophysiology
A halogenated anesthetic agent alone may
trigger an episode of MH (Table 52–2). In many of the early reported cases, both succinylcholine and a
haloge-nated anesthetic agent were used. However, succi-nylcholine is less
frequently used in modern practice, and about half of the cases in the past
decade were associated with volatile anesthetics as the only triggering agents. Nearly 50% of patients
who experience an episode of MH have had at leastone previous uneventful
exposure to anesthesia dur-ing which they received a recognized triggering
agent. Why MH fails to occur after every exposure totriggering agent is
unclear. Investigations into the biochemical causes of MH reveal an
uncontrolled increase in intracellular calcium in skeletal muscle. The sudden
release of calcium from sarcoplasmic reticulum removes the inhibition of
troponin, resulting in sustained muscle contraction. Markedly increased
adenosine triphosphatase activity results in an uncontrolled increase in
aerobic and anaerobic metabolism. The hypermetabolic state markedly increases
oxygen consumption and CO 2 production, producing severe lactic acidosis and hyperthermia.
One early focus of investigations into the
mech-anisms of MH has been the gene for the ryanodine (Ryr 1) receptor, located on chromosome 19. Ryr1 is

an ion channel responsible for calcium release from the sarcoplasmic
reticulum and it plays an impor-tant role in muscle depolarization. Subsequent
reports linked MH with mutations involving the sodium channel on chromosome 17.
An autosomal recessive form of MH has been associated with the King–Denborough
syndrome. Most patients with an episode of MH have a history of relatives with similar episode or with an abnormal halothane– caffeine contracture test
. The complex-ity of genetic inheritance patterns in families reflects the fact
that MH can be caused by mutations of one or more genes on more than one
chromosome. To date genetic studies in humans have revealed at least five
different chromosomes and more than 180 indi-vidual mutations associated with
MH. Genetic test-ing, although available, currently screens for less than 20%
of recognized mutations. A patient with a bona fide clinical history of MH has
about a 30–50% chance of testing positive.
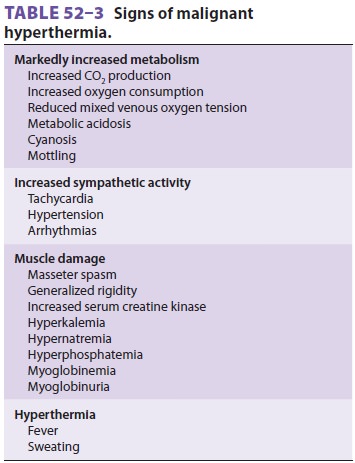
Clinical Manifestations
The earliest signs of MH during anesthesia
are succinylcholine-induced masseter musclerigidity (MMR) or other muscle
rigidity, tachycar-dia, and hypercarbia (due to increased CO2 pro-duction) (Table 52–3). Two or more of these signs greatly increase the likelihood of MH.
Tachypnea is prominent when muscle relaxants are not used. Overactivity of the
sympathetic nervous system produces tachycardia, arrhythmias, hypertension, and
mottled cyanosis. Hyperthermia may be a late sign, but when it occurs, core
temperature can rise as much as 1°C every 5 min. Generalized muscle rigidity is not consistently present.
Hypertension may be rapidly followed by hypotension if cardiac depression
occurs. Dark-colored urine reflects myo-globinemia and myoglobinuria.
Laboratory testing typically reveals mixed metabolic and respiratory
acidosis with a marked base deficit, hyperkalemia, hypermagnesemia, and reduced
mixed-venous oxygen saturation. Some case reports describe isolated respiratory
acido-sis early in the course of an episode of MH. Serum ionized calcium
concentration is variable: it may initially increase before a later decrease.
Patients typically have increased serum myoglobin, creatine
kinase (CK), lactic dehydrogenase, and aldolase lev-els. When peak serum
CK levels (usually 12–18 h after anesthesia) exceed 20,000 IU/L the diagnosis
is strongly suspected. It should be noted that succi-nylcholine administration
to some normal patients without MH may cause serum myoglobin and CK levels to
increase markedly.
Much of the problem in diagnosing MH arises from its variable
presentation. Fever is an inconsis-tent and often late-presenting sign. An
unantici-pated doubling or tripling of end-tidal CO2 (in the
absence of a ventilatory change) is one of the earliest and most sensitive
indicators of MH. If the patient survives the first few minutes, acute kidney
failure and disseminated intravascular coagulation (DIC) can rapidly ensue.
Other complications of MH include cerebral edema with seizures and hepatic
failure. Most MH deaths are due to DIC and organ failure due to delayed or no
treatment with dantrolene.
Susceptibility to MH is increased in sev-eral
musculoskeletal diseases. These includecentral-core disease, multi-minicore
myopathy, and King–Denborough syndrome. The latter syndrome is seen primarily
in young boys who exhibit short stature, mental retardation, cryptorchidism,
kypho-scoliosis, pectus deformity, slanted eyes, low-set ears, webbed neck, and
winged scapulae. Duchenne’s and other muscular dystrophies, nonspecific
myopathies, heat stroke, and osteogenesis imperfecta have been associated with
MH-like symptoms in some reports; however, their association with MH is
controversial. Other possible clues to
susceptibility include a fam-ily history of anesthetic complications, or a
history of unexplained fevers or muscular cramps. There are several reports of
MH episodes occurring in patients with a history of exercise-induced
rhabdomyolysis. Prior uneventful anesthesia procedures and absence of a
positive family history are notoriously unreli-able predictors of lack of
susceptibility to MH. Any patient who develops MMR during induction of
anesthesia should be considered potentially suscep-tible to MH.
Intraoperative Considerations
Treatment of an MH episode is directed at
terminating the episode and treating complications such as hyperthermia and
acidosis. The mortality rate for MH, even with prompt treat-ment, ranges from 5%
to 30%. Table 52-4 illustrates

a standard protocol for management of
MH. First and most importantly, the triggering agent must be stopped and
dantrolene must be given immediately.
A. Acute Treatment Measures
Volatile agents and succinylcholine must be dis-continued immediately.
Even trace amounts of anesthetics absorbed by soda lime, breathing tubes, and
breathing bags may be detrimental. The patient should be hyperventilated with
100% oxygen to minimize the effects of uncontrolled CO 2
produc-tion and increased oxygen consumption.
B. Dantrolene Therapy
The mainstay of therapy for MH is immediate administration of intravenous dantrolene. Dan-trolene, a hydantoin
derivative, directly interferes with muscle
contraction by binding the Ryr1 receptor channel and inhibiting calcium ion release from the
sarcoplasmic reticulum. The dose is 2.5 mg/kg intravenously every 5 min until
the episode is terminated (upper limit, 10 mg/kg). Dantrolene is packaged as 20
mg of lyophilized powder to be dis-solved in 60 mL of sterile water. Depending
on the dose required and drug formulation used, reconsti-tution can be time
consuming. An
assistant may beneeded. A new formulation
isavailable that recon-stitutes in about one third the time (20 versus 86 s)
required for the older formulation. The effective half-life of dantrolene is
about 6 h.
After initial control of symptoms, 1 mg/kg of
dantrolene intravenously is recommended every 6 h for 24–48 h to prevent relapse
(MH can recur within 24 h of an initial episode). Dantrolene is a relatively
safe drug that is also used to decrease temperature in patients with thyroid
“storm” and neuroleptic malignant syndrome. Although its use in chronic therapy
for spastic disorders has been associated with hepatic dysfunction, the most
serious com-plication following acute administration is general-ized muscle
weakness that may result in respiratory insufficiency or aspiration pneumonia.
Dantrolene can cause phlebitis in small peripheral veins and should be given
through a central venous line if one is available. The safety and efficacy of
dantrolene therapy mandate its immediate use in this poten-tially
life-threatening situation. Following adminis-tration of dantrolene, most patients
revert to normalacid–base status promptly and no further pharma-cological treatment is
necessary.
C. Correction of Acid–Base/ Electrolyte Imbalances
Persisting metabolic acidosis should be
treated with intravenous sodium bicarbonate, recogniz-ing that this treatment
will worsen the hypercarbia. Hyperkalemia should be treated with glucose,
insu-lin, and diuresis. There is no useful role for intrave-nous calcium in
this setting. Antiarrhythmic agents, vasopressors, and inotropes should be
administered, if indicated. Calcium channel blockers should not be given to
patients receiving dantrolene because this combination appears to promote
hyperkalemia. Furosemide may be used to establish diuresis and prevent acute
kidney failure, which may develop as a consequence of myoglobinuria. Dantrolene
contains a considerable amount of mannitol (3 g per 20-mg bottle); thus
furosemide or bumetanide should be used in preference to mannitol for diuresis.
D. Cooling the Patient
If fever is present, cooling measures should be insti-tuted immediately.
Surface cooling with ice packs over major arteries, cold air convection, and
cooling blankets are used. Iced saline lavage of the stomach and any open body
cavities (eg, in patients under-going abdominal surgery) should also be
instituted. Use of hypothermic cardiopulmonary bypass may be appropriate if
other measures fail.
E. Management of the Patient with Isolated Masseter Muscle Spasm
MMR, or trismus, is a forceful contraction of the jaw musculature that
prevents full mouth open-ing. This contrasts with incomplete jaw relaxation,
which is a fairly common finding. Both myotonia and MH can cause masseter
spasm. The two disor-ders can be differentiated by the medical history,
neurological examination, and electromyography. The historical incidence of MMR
following admin-istration of succinylcholine with halothane in pedi-atric
patients at some medical centers was higher than 1%. Isolated MMR occurs in
only 15–30% of true MH episodes. Moreover, less than 50% of patients in whom
MMR develops prove to be sus-ceptible to MH by muscle testing. In the past,
theconsensus of clinicians was to assume that any occurrence of MMR was
diagnostic of MH and to postpone elective surgery. However, if there is no
other sign of MH, and if monitoring and treatment capabilities are readily
available, many anesthesi-ologists now advocate allowing surgery to continue
using safe (nontriggering) anesthetic agents. Serum CK levels should be
followed for 24 h after an epi-sode of MMR, because an elevation of this enzyme
may indicate an underlying myopathy.
Postoperative Considerations
A. Confirmation of the Diagnosis
Patients who have survived an unequivocal
episode of MH are considered susceptible; in these patients a muscle biopsy
need not be performed for diagnosis. If the diagnosis remains in doubt
postoperatively,fresh biopsy specimen of living skeletal muscle is obtained and exposed
to a caffeine, halothane, or combination caffeine–halothane bath. The
halo-thane–caffeine contracture test may have a 10–20% false-positive rate, but
the false-negative rate is close to zero. Because of the relative complexity of
this test, only a few centers worldwide perform it. If the halothane–caffeine
contracture test is posi-tive, genetic counseling and testing of family
mem-bers are appropriate. Baseline CK may be elevated chronically in 50–70% of
people at risk for MH, but the only reliable way to diagnose MH susceptibility
is by muscle testing.
Both European and North American MH regis-tries have been established to
help physicians iden-tify and treat patients with suspected MH, as well as
provide standardization between testing centers. The Malignant Hyperthermia
Association of the United States (MHAUS, telephone 1-800-986-4287) operates a
24-hour hotline (1-800-644-9737) and a web site (http://www.mhaus.org).
1.
Differential diagnosis—Severaldisorders
maysuperficially resemble MH (Table 52–5). However, MH is associated with greater degrees of metabolic acidosis
and venous desaturation than any of these other conditions. In current
practice, the most common condition confused with MH is hypercar-bia from CO2 insufflation for laparoscopy, with or
without subcutaneous emphysema. This condition can result in an unexpected
increase in end-tidal

CO2 with accompanying tachycardia. Surgery and anesthesia can precipitate
thyroid storm in undiag-nosed or poorly controlled hyperthyroid patients. The
signs of thyroid storm include tachycardia, tachyarrhythmias (particularly
atrial f briillation), hyperthermia (often ≥40°C), hypotension, and in some cases
congestive heart failure. In contrast to MH, hypokalemia is very common. Also
unlike the typical intraoperative presentation of MH, thy-roid storm generally
develops postoperatively . Pheochromocytoma is associated with dramatic
increases in heart rate and blood pressure but not with an increase in CO2
production, endtidal CO 2, or temperature . Car-diac
arrhythmias or ischemia may also be prominent. Rarely such patients may have
hyperthermia (>38°C), which is generally thought
to be due to increased heat production from catecholamine-mediated increases in
metabolic rate together with decreased heat elimination from intense
vasocon-striction. Sepsis shares several characteristics with MH, including
fever, tachypnea, tachycardia, and metabolic acidosis . Sepsis can be difficult
to diagnose if there is no obvious primary site of infection.
Less commonly, drug-induced hyperthermia may
be encountered in the perioperative period. In these cases, the drugs appear to
markedly increase serotonin activity in the brain, causing hyperther-mia,
confusion, shivering, diaphoresis, hyperreflexia, and myoclonus. Drug
combinations associated with this “serotonin syndrome” include monoamine oxidase
inhibitors (MAOIs) and meperidine, and MAOIs and
selective serotonin reuptake inhibitors (SSRIs). Hyperthermia can also be
caused by some illicit drugs, including 3,4-methylenedioxymetham-phetamine
(MDMA or “ecstasy’), “crack” cocaine, amphetamines, phencyclidine (PCP), and
lysergic acid diethylamine (LSD).
Iatrogenic hyperthermia is not uncommon, particularly in pediatric
patients. Common sources of excessive heat in the operating room include
humidifiers on ventilators, warming blankets, heat lamps, and increased ambient
temperature. Injuries to the brainstem, hypothalamus, or nearby regions can be
associated with marked hyperthermia.
2. Neuroleptic
malignant syndrome (NMS)—This syndrome is
characterized by hyperthermia, muscle rigidity with extrapyramidal signs (dyskine-sia),
altered consciousness, and autonomic lability in patients receiving
antidopaminergic agents. The syndrome is caused by an imbalance of
neurotrans-mitters in the central nervous system. It can occur either during
drug therapy with antidopaminergic agents (eg, phenothiazines, butyrophenones,
thio-xanthenes, or metoclopramide) or less commonly following the withdrawal of
dopaminergic agonists (levodopa or amantadine) in patients with Parkin-son’s
disease. Thus, it appears to involve abnormal central dopaminergic activity, as
opposed to the al-tered peripheral calcium release seen in MH. These differing
mechanisms probably explain why nonde-polarizing relaxants reverse the rigidity
of NMS, but not the rigidity associated with MH.
NMS does not appear to be inherited and
typi-cally takes hours to weeks to develop; the majority of episodes develop
within 2 weeks of a dose adjust-ment. Hyperthermia generally tends to be mild,
and appears to be proportional to the amount of rigidity. Autonomic dysfunction
results in tachycardia, labile blood pressure, diaphoresis, increased
secretions, and urinary incontinence. Muscle rigidity can pro-duce dyspnea and
respiratory distress and, together with the increased secretions, can promote
aspiration pneumonia. CK levels are typically elevated; some patients may
develop rhabdomyolysis resulting in myoglobinemia, myoglobinuria, and kidney
failure.
Mild forms of NMS promptly resolve after withdrawal of the causative
drug (or reinstitution of antiparkinsonian therapy).
Initial treatment of more severe forms of NMS should include oxygen therapy and
endotracheal intubation for respiratory distress or altered consciousness.
Marked muscle rigidity can be controlled with muscle paralysis, dantrolene, or
a dopaminergic agonist (amantadine, bromocriptine, or levodopa), depending on
the severity and acuity of the syndrome. Resolution of the muscle rigidity
usually decreases body temperature.
This syndrome is considered a separate entity from MH; nevertheless some
clinicians believe that NMS may predispose patients to MH and recom-mend that
patients with NMS should not receive succinylcholine or a volatile anesthetic.
In contrast to patients with NMS, patients susceptible to MH can safely receive
phenothiazines.
B. Prophylaxis, Postanesthesia Care, and Discharge
Propofol, etomidate, benzodiazepines,
ket-amine, thiopental, methohexital, opiates,droperidol, nitrous oxide,
nondepolarizing muscle relaxants, and all local anesthetics are nontrigger-ing
agents that are safe for use in MH-susceptible patients. An adequate supply of
dantrolene should always be available wherever general anesthesia is provided.
Prophylactic administration of intrave-nous dantrolene to susceptible patients
is not nec-essary if a nontriggering anesthetic is administered.
For MH-susceptible patients, the consensus is
that the vaporizers should be removed from the anesthesia workstation (or fixed
in an “off ” position) and the machine should be flushed with 10 L/min of fresh
gas (air or oxygen) for at least 5 min. This step should reduce concentrations
of volatile anesthetics to less than 1 part per million. Additionally, the CO2 absorbent and circle system (or other
anesthetic cir-cuit), hoses should be changed.
MH-susceptible patients who have under-gone
an uneventful procedure with a nontrigger-ing anesthetic can be discharged from
the PACU or ambulatory surgery unit when they meet standard criteria. There are
no reported cases of MH-susceptible patients experiencing MH after receiving a
nontriggering anesthetic during unevent-ful surgery.
Related Topics