Chapter: Pharmaceutical Drug Analysis: Liquid-Liquid Extraction
Liquid-Liquid Extraction: Theory
THEORY
The behavioural pattern of two immiscible solvents, say ‘a’ and ‘b’, is essentially nonideal with respect to one another. Now, if a
third substance is made to dissolve in a two-phase mixture of the solvents (i.e., ‘a’ and ‘b’), it may
behave ideally in either phases provided its concentration in each individual
phase is approximately small. Therefore, under these prevailing experimental
parameters the ratio of the mole fractions of the solute in the two respective
immiscible phases (‘a’ and ‘b’) is found to be a constant which is
absolutely independent of the quantity of solute present. It is termed as the Nernst Distribution Law or the Partition Law and may be expressed as
follows :
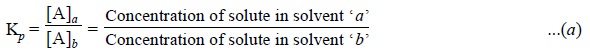
where, [A]a = Mole
fraction of solute A in Phase ‘a’
[B]b
= Mole fraction of solute B in Phase ‘b’,
and
Kp =
A constant.
The constant (Kp)
is also known as the distribution
coefficient or the partition
coefficient. Interest-ingly, this particular relation [Eq. (a)] was originally derived for ideal
solutions only, but it caters for a fairly good description of the behavioural
pattern of a number of real-extraction-systems encountered in the analy-sis of
pharmaceutical substances. However, the Partition
Law offers the following two
limitations, namely :
(a) It is not
thermodynamically rigorous i.e., it
takes no cognizance of the activities of the different species. In other words,
it is solely applicable to very dilute solutions in which case the ratio of the
activities almost approaches unity, and
(b) It does not
hold good when the distributing substances encounters association or
distribution in either phases (i.e.,
‘a’ and ‘b’).
Consequently, a more rigorous treatment particularly
specifies Kp as the ratio
of the activities of the substance (A) in the two solvents instead of their
concentrations. Hence, for dilute solutions, at a specified constant pressure
and temperature, the mole fraction of a solute is directly proportional to its
concentration in molarity or mass per unit volume ; which implies that these
may be employed instead of mole-fraction in Eq. (a).
Thus, the Partition
Coefficient Kp is also
given by the following expression :
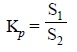 ..............................(b)
..............................(b)
where, S1 = Solubility of substance A in
solvent ‘a’, and
S2 = Solubility of substance A in solvent ‘b’.
Adequate precaution and care must be exercised in
determining partition coefficients based on the solubility data as S1
is not the solubility of substance ‘A’ in pure Solvent ‘a’, but rather the solubility in Solvent ‘a’ saturated with Solvent ‘b’.
Example : In
order to determine the exact partition coefficient of substance ‘A’ between
water and ethyl acetate, the appropriate solubilities would be those of
the substance ‘A’ in 3.3% ethyl acetate in water (composition of the ‘aqueous’
layer) and 8.7% in water in ethyl acetate (composition of the ‘ester’ layer).
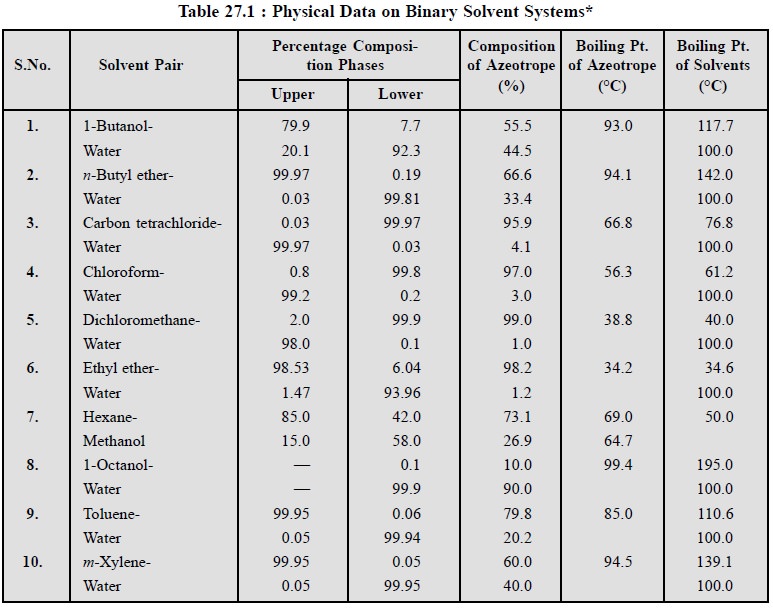
Likewise, the following Table 27.1, records the mutual
solubilities of a few typical solvent pairs that are used frequently for liquid-liquid extraction procedures.
In liquid-liquid extractions the following two aspects are very crucial and
important, namely :
(a) Error due
to the Volume Change, and
(b)
Effectiveness of an Extraction.
These two
aspects shall be discussed briefly at this juncture.
1. ERROR DUE TO THE VOLUME CHANGE
In a situation wherein two immiscible solvents are
employed in an extraction, the volumes of the two individual phases after
attainment of equilibrium may be appreciably different in comparison to the
initial volumes of the solvents used. Therefore, a number of procedures have
been adopted to avoid ‘error due to the volume change’ incurred thereby,
namely :
(i) Measure the
volume of the phase employed for the analysis and incorporate this volume in
the calculations,
(ii) Separate
the phase quantitatively and subsequently dilute to a known volume,
(iii) Separate
the phase quantitatively and make use of the entire volume in the remaining
steps of the ongoing analysis, and
(iv) Carry a
marker substance through the extraction to automatically compensate for volume
changes.
However, the latter procedure finds its abundant use in
chromatographic methods of analysis.
2. EFFECTIVENESS OF AN EXTRACTION
Based on the appropriate partition coefficient of an
immiscible solvent pair it is possible to calculate the ‘effectiveness of an
extraction’.
Let us assume that ‘x’
moles of solute present initially in a volume V2 of Solvent ‘b’. Now, this particular sample
undergoes extraction with a volume V1 of Solvent ‘a’ and subsequently ‘y’ moles of com-pound are left in V2
at equilibrium.
Substituting these values in Eq. (a) and using molarity instead of mole fraction, we have :

after simplifying and rearranging :
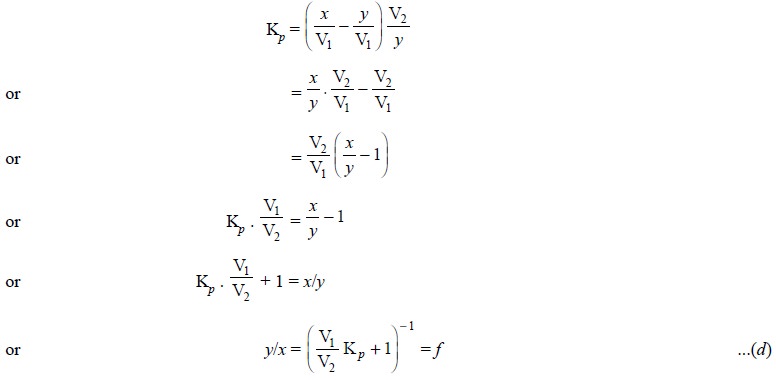
where, f = Fraction not
extracted.
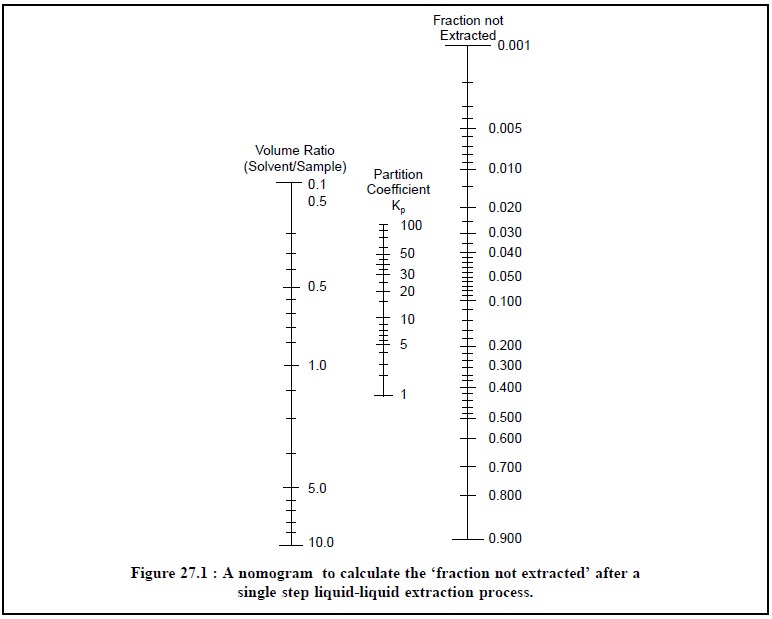
Figure 27.1, represents the nomogram from which the
unextracted fraction for various values of V2/V1 and K
may be obtained.
From Eqs. (d)
it is quite evident that the fraction extracted is absolutely independent of
the initial solute concentration. Hence, the fraction left unextracted after ‘n’ extraction may be given by the
following expression:
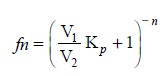 ..................................(e)
..................................(e)
assuming that the same volumes of solvents have been used
for each extraction.
Form Eq. (e) it
is distinctly obvious that a series of several extractions would definitely
prove to be more efficacious than one single extraction using the same total
volume of solvent.
From figure 27.1, the following steps may be adopted in
order to determine the percentage of the analyte left in the sample after a
single extraction :
(i)
Hold a straight edge in such a manner that it is made to
pass though the point on the ‘left scale’ which exactly corresponds to that
ratio of extracting solvent to sample solvent volume and through the point
representing the partition coefficient (Kp)
on the ‘middle scale’, and
(ii)
The percentage of the ‘analyte’, left behind in the sample solvent after a single
extraction is given by the intersection of the straight-edge with the ‘right scale’.
Related Topics