Chapter: Pharmaceutical Drug Analysis: Liquid-Liquid Extraction
Factors Influencing Solvent Extraction
FACTORS INFLUENCING SOLVENT EXTRACTION
A number of cardinal factors exert a positive influence
on the phenomenon of solvent extraction, namely :
(a) Effect of
temperature and inert solutes,
(b) Effect of
pH on extraction,
(c) Effect of
ion-pair formation, and
(d) Effect of
synergistic extraction.
These factors shall be discussed briefly below :
1. EFFECT OF TEMPERATURE AND INERT SOLUTES
The physical as well as chemical interactions of a solute
is capable of changing its apparent partition coefficient between a pair of
solvents. Therefore, it is absolutely necessary to take this into consideration
while selecting an appropriate extraction-system. Craig and Craig* have
advocated that the partition coefficients are normally not sensitive to
temperature when the two solvents in question are more or less immiscible and
also the concentrations are fairly low in both the phases. Thus, the effect of
temperature on the partition coefficient may be estimated conveniently from its
effect on the solubilities of the substance in the two respective solvents. By
substituting the solubilities (e.g.,
S1 and S2) in Eq. (b)
it is possible to estimate K.
The effect of inert solutes, such as : calcium chloride,
magnesium chloride and sucrose, can also be employed judiciously and
efficaciously in the development of solutions to difficult extraction problems
by allowing efficient extractions from the water into such solvents as acetone,
ethanol and methanol that are found to be completely miscible with water in the
absence of salt. Matkovitch and Cristian* found the above three inert solutes
to be the best agents for salting acetone out of water. It has been observed
that the acetone layer that separated from a saturated aqueous solution of CaCl2
exclusively contained 0.32 ± 0.01% water (v/v) and 212 ppm
salt (w/w) at equilibrium.
2. EFFECT OF pH ON EXTRACTION
Generally, it has been found that the organic acids and
bases do exist in aqueous solution as equilib-rium mixtures of their respective
neutral as well as ionic forms. Thus, these neutral and ionic forms may not
have the same identical partition coefficients in a second solvent ; therefore,
the quantity of a substance being extracted solely depends upon the position of
the acid-base equilibrium and ultimately upon the pH of the resulting solution.
Hence, extraction coefficient (E) may be defined as the ratio of the
concentrations of the substance in all its forms in the two respective phases
in the presence of equilibria ; and it can be expressed as follows :
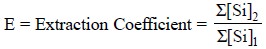 ................................(i)
................................(i)
where, Σ[Si]2 = The sum
total of all forms of the compound in Phase-‘2’, and
Σ[Si]1 =
The sum total of all forms of the compound in Phase ‘1’.
In fact, the actual effect of the equilibrium on the
extraction may be shown by determining the extrac-tion coefficient for the
system :
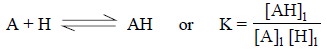 ............................(ii)
............................(ii)
where, A = Extract with partition coefficient Kp, A and
AH = Extract with partition coefficient Kp, AH
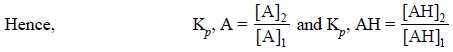 ........................(iii)
........................(iii)
Therefore, for this particular system the efficiency
coefficient E may be expressed as follows :
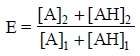 .............................(iv)
.............................(iv)
Now, substituting Eq. (ii) and Eq. (iii) into
Eq. (iv) and subsequently
simplifying, we shall get :
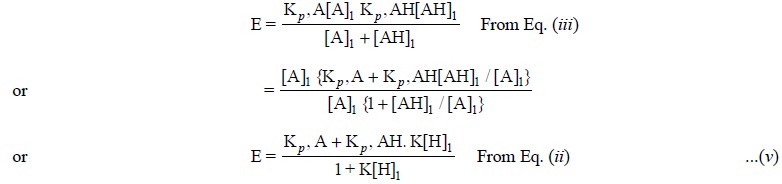
From Eq. (v) it
is quite evident that E approaches Kp,
A as K[H]1 becomes small and Kp,
AH as K[H]1 becomes large.
Now, assuming that only A extracts (i.e., A being a neutral organic base and AH the conjugate acid),
Eq. (v) may be
expressed as :
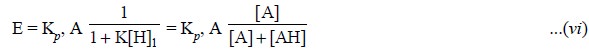
The following inferences may be arrived at on the basis
of Eq. (vi), namely :
(a) Extraction
coefficient (E) is just the partition coefficient times the fraction of the
analyte which is present in the extractable form,
(b) Under a
given set of experimental parameters the ultimate effect of the ‘equilibrium’
shall be to reduce the amount extracted, and
(c) Forcibly
shifting the ‘equilibrium’ toward the extractable species by adjusting the pH
helps to minimise the effect of the equilibrium thereby rendering E almost
equal to Kp, A.
In conclusion, it may be observed that the pH for an ‘extraction system’ must be selected in
such a fashion so that the maximum quantum of the analyte is present in the
extractable form, that obviously suggests that the analyte should always be in
the form of either a free base or a free acid. From the actual practical
experience it has been noticed that a good-working range lies between 95 to 97%
present in the extractable form.
3. EFFECT OF ION-PAIR FORMATION
Ion-pair formation needs its due recognition
because it very often gives rise to unexpected extrac-tions. In true sense,
ion-pair may be regarded as a close association of an anion and cation, and
therefore, it usually takes place either in a polar or a non-polar solvent. In
reality, the ion-pairs are invariably formed by virtue of the union between
comparatively large organic anions and (much smaller) cations. Interestingly,
the resulting ion-pairs have been found to show their appreciable solubility in
polar solvents ; and hence, these species may be extracted conveniently under
such experimental parameters where neither individual compo-nent ion could.
A few vital criteria towards the formation of an improved
aqueous extractable ionic species are, namely :
·
Formation of a
neutral metal-chelate complex or by ion association, and
·
Creation of larger and more hydrophobic molecular
species.
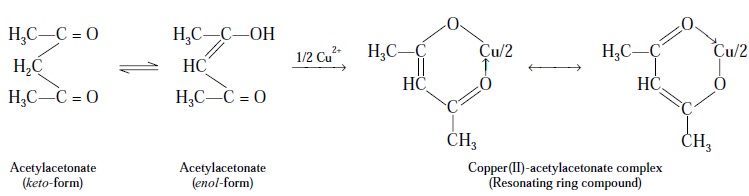
A few typical examples shall be discussed here to explain
the chelate-formation :
Example
1 : Cu2+ with ‘acetylacetonate’ forms a fairly stable ring
compound :
Example 2 : Iron (III)
‘cupferrate’ gives rise to a stable ring
compounds as shown below :
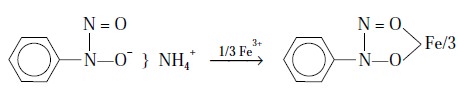
Example 3 : Sulphonic acids rapidly pair with a plethora
of
‘protonated amines’ to form
an easily extractable complex
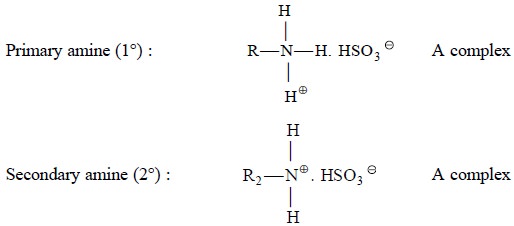
Example 4 : Cl– ion
serves
as an ‘appropriate anion’ that favourably combines with many aromatic amines
and alkaloids which may ultimately be extracted from the corresponding aqueous
solutions into chloroform as their respective chlorides*.
4. EFFECT OF SYNERGISTIC EXTRACTION
Synergism : It may be defined as ‘the process whereby two different
reagents when employed together are capable of extracting a metal ion with a
distinct and marked efficiency, in comparison to a condition when the same two
reagents are used individually’.
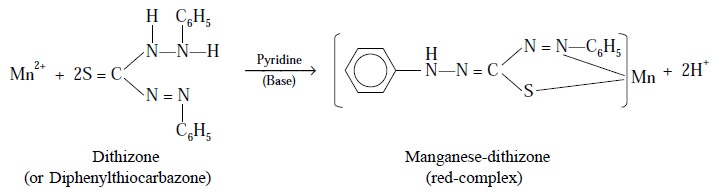
Example : (i) : Complexation of Mn2+ with dithizone and pyridine :
It has been observed that the complex formed by Mn2+
with dithizone alone is of no practical analyti-cal utility because of the fact
that it undergoes decomposition very quickly. However, the addition of a base,
such as : pyridine into the Mn2+ plus dithizone complex yields a
red-complex, which is fairly stable to oxidation and light; and, therefore,
forms the basis for a very sensitive photometric
method employed in estimating trace amounts of Mn2+.
Equation : Following is the chemical
reaction of the above complex formation :
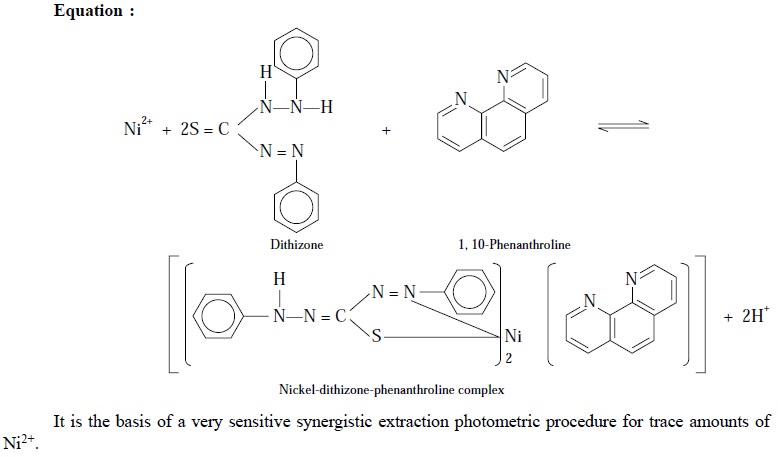
(ii) Complexation of Ni2+ with dithizone and 1,
10-phenanthrolone :
Noticeably, the reaction of Ni2+ with
dithizone is quite slow and sluggish. Nevertheless, this slow reaction is
significantly accelerated by the addition of nitrogen-containing bases like 1,
10-phenanthroline. The resulting complex may be represented by the following
equation :
Related Topics