Chapter: Medical Immunology: Immune System Modulators
Immunosuppressive Drugs: Pharmacological and Immunological Aspects
Immunosuppressive Drugs: Pharmacological and Immunological Aspects
A variety of drugs, ranging from glucocorticoids to cytotoxic drugs, have been used for the purpose of suppressing undesirable immune responses. While many of these drugs are loosely termed “immunosuppressive,” they differ widely in their mechanisms of action, toxicity, and efficacy. The exact mechanisms of action of immunosuppressive drugs are difficult to determine, partly because the physiology of the immune response has not yet been completely elucidated. The targets of immunosuppressive therapies are rather diverse and, depending on the agents, may include phagocytosis and antigen-processing by macrophages; antigen recognition by lymphocytes; proliferation and/or differentiation of lymphocytes; production of cytokines; immune effector mechanisms, including the pro-duction and release of cytotoxic leukocytes, antibodies, and/or delayed hypersensitivity mediators.
1. Hormones
The major agents in this group of substances are the glucocorticoid hormones of the adrenal complex cortex and their synthetic analogs (glucocorticoids or corticosteroids, such as prednisone and methylprednisolone). The mechanisms of actions of glucocorticoids are still being defined, but they can be divided into three major effects.
Induction of Apoptosis.At certain dosage levels, treatment with glucocorticoidsmay produce a rapid and profound lymphopenia. This is particularly true in cases of lym-phocytic leukemia, and it is a consequence of the induction of apoptosis. The molecular mechanism of glucocorticoid-induced apoptosis hinges on the activation of an endonucle-ase, which is normally inactive due to its association to a protein. This effect, like all other cellular effects of glucocorticoids, including the induction of apoptosis, requires associa-tion with a glucocorticoid cytoplasmic receptor. The glucocorticoid-receptor complex is translocated to the nucleus, where it binds to regulatory DNA sequences (glucocorticoid-responsive elements). At the genetic level, two possibilities have been suggested to account for the enhancement of apoptotic processes based on experimental observations:
1. Downregulation of the synthesis of the protein that inactivates the endonucle-ase responsible for DNA breakdown
2. Activation of an ICE-like protease that degrades the endonuclease-inactivating protein
Downregulation of Cytokine Synthesis.The administration of glucocorticoids isfollowed by a general downregulation of cytokine synthesis. This effect is secondary to the inhibition of nuclear binding proteins that activate the expression of cytokine genes. Two mechanisms (not mutually exclusive) have been proposed (Fig. 24.1).
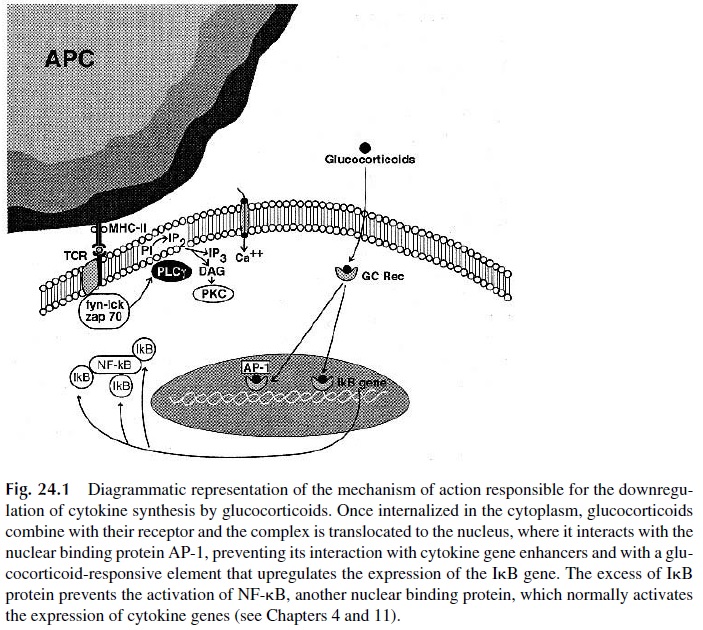
1. After combining with the glucocorticoid cytoplasmic receptor, the glucocorti-coid-receptor complex is translocated to the nucleus, where it prevents the as-sociation of AP-1 with promoter sequences controlling the expression of cy-tokine genes.
2. The translocated glucocorticoid-receptor complex binds to the promoter of the inhibitory protein that regulates the activity of NF Bk (IkB;) and induces its expression. The synthesis of abnormally high levels of IkB results in the inactivation of NFkB, thus neutralizing a second nuclear-binding protein that enhances the expression of cytokine genes.
Anti-Inflammatory Effects.The anti-inflammatory effect of glucocorticoids isprobably the most significant from the pharmacological point of view. Several actions of glucocorticoids combine to induce this anti-inflammatory effect:
1. Downregulation of the synthesis of pro-inflammatory cytokines.
2. Reduced expression of CAMs on the vessel wall, partly as a consequence of the downregulation of cytokine synthesis (pro-inflammatory cytokines upregulate CAM expression) and partly as a consequence of a direct downregulation of the expression of the genes encoding those molecules. The modulation of CAM ex-pression has a marked effect on leukocyte traffic. Neutrophils and T lympho-cytes are predominantly affected and remain sequestered on the bone marrow and lymph nodes, impairing their ability to generate both specific immune re-sponses and nonspecific inflammatory responses.
3. The synthesis of phospholipase A2 is downregulated due to the binding of the glucocorticoid-receptor complex to a DNA glucocorticoid-responsive element that has a negative effect on the expression of the phospholipase A2 gene. Con-sequently, the synthesis of leukotrienes, prostaglandins, and platelet-activating factor is also downregulated.
4. The nitric oxide synthase gene is also downregulated; thus, the release of nitric oxide is reduced, eliminating its vasodilator effect.
5. In contrast, glucocorticoids upregulate the expression of lipocortin-1, a protein that has anti-inflammatory effects in part due to its ability to inactivate pre-formed phospholipase A2.
Nonsteroidal Anti-inflammatory Drugs.Nonsteroidal anti-inflammatory drugs(NSAIDs) are totally unrelated to the corticosteroid family but share certain anti-inflamma-tory properties. The anti-inflammatory potency of the NSAID family appears to directly cor-relate with their ability to inhibit prostaglandin and thromboxane synthesis from arachldonic acid by inhibiting the enzyme cyclooxygenase (COX). There are two isoforms of the COX en-zyme. COX-1 is constitutively expressed in most cells, while COX-2 is induced in inflamed tissue only. The anti-inflammatory properties of both selective and nonselective NSAIDs ap-pear to be related to their inhibition of COX-2. Some of the adverse effects of the nonselective NSAIDs (e.g., gastrointestinal and renal toxicity) may be related to the inhibition of COX-1.
NSAIDs include a large number of prescription and over-the-counter (OTC) drugs. Common examples of the nonselective COX-1 and -2 inhibitors include aspirin, ibuprofen, indomethacin, naproxen, and ketorolac. There are currently only two FDA-approved se-lective COX-2 inhibitors, celecoxib and rofecoxib. NSAIDs do not appear to modulate cy-tokine release or to alter T- and B-lymphocyte cell trafficking, and thus have virtually no immunosuppressive effects.
2. Alkylating Agents and Antimetabolites
Several commonly used immunosuppressants fit into this category. They can be generically classified into two large groups: cell-cycle specific and cell-cycle nonspecific. The choice between the two types of immunosupressants is based both on clinical experience and on their characteristics. For example, for the treatment of autoimmune diseases that form ab-normal immune humoral- and cell-mediated immune responses, the goal is to turn off the proliferating autoreactive lymphocytes, and cell-cycle–specific agents with the ability to block DNA synthesis are utilized. On the other hand, plasma cell malignancies are difficult to treat with cell-cycle–specific agents, because the majority of antibody-producing plasma cells are not in cycle, but rather in a prolonged G1 or “G0” state. In this case, cell-cycle–non-specific agents are most likely to be effective.
Cell-cycle–specific agents cause cell death by interfering with different parts of the cell division cycle. This group includes the antimetabolites methotrexate, azathioprine, and 6-mercaptopurine (6-MP), which appear to act only on cells in the S-phase, when DNA is actively synthesized. Two newer compounds, mycophenolate mofetil and leflunomide, are also included in this group.
Methotrexate, a folic acid analog, binds to and inhibits dihydrofolate reductase, thus blocking the formation of the DNA nucleotide thymidine. It is most active against cells in the S-phase of the cell cycle. T lymphocytes, after appropriate antigen stimulation, begin to proliferate and enter the S-phase of the cell cycle. Therefore, methotrexate is used to block lymphocyte proliferation. Unfortunately, methotrexate also targets other rapidly di-viding cells (e.g., bone marrow, gastrointestinal tract, hair follicles), leading to its common side effects (agranulocytosis and other consequences of bone marrow suppression, diarrhea and other gastrointestinal symptoms secondary to mucosal damage, and alopecia). Methotrexate is commonly used to treat NSAID-refractory rheumatoid arthritis and malig-nancies (e.g., breast cancer and acute lymphoblastic leukemia).
Azathioprine has been used in solid organ transplantation since 1963. After absorp-tion, azathioprine is metabolized by hepatic and RBC gluthathione to 6-mercaptopurine. In-tracellularly, 6-MP is converted to form thiopurine ribonucleosides and nucleotides that in-hibit purine synthesis by both the de novo pathway and salvage pathway and thereby inhibit the proliferation of T and B lymphocytes. Azathioprine was commonly used with cy-closporine and prednisone as the “standard” regimen for prevention of solid organ trans-plant rejection. More recently mycophenolate has been taking its place in these combina-tions . In addition, azathioprine is used to treat Crohn’s disease. 6-MP is mainly used in combination with methotrexate as part of maintenance therapy for acute lym-phoblastic leukemia in children.
Mycophenolate mofetil (MMF, Cellcept) also inhibits lymphocyte proliferation. It acts as a reversible inhibitor of inosine monophosphate dehydrogenase, thus interfering with the de novo pathway of guanine nucleotide synthesis and subsequent DNA replica-tion. T and B lymphocytes are highly dependent on the de novo pathway for the generation of guanosine nucleotides whereas other cells can use the salvage pathway. Thus, my-cophenolate affects T and B lymphocytes with some degree of selectivity over other types of cells. Mycophenolate is used clinically in prophylaxis of organ rejection in solid organ transplantation.
Leflunomide, recently approved by the U.S. FDA, also hinders the proliferation of lymphocytes by inhibiting a different enzyme, dihydro-orotate dehydrogenase. This is a key enzyme in de novo pyrimidine synthesis, and activated T lymphocytes primarily syn-thesize pyrimidines by this de novo pathway. In three randomized trials, leflunomide ap-pears to exhibit efficacy similar to sulfasalazine (a NSAID) and methotrexate in patients with active rheumatoid arthritis.
Alkylating agents such as cyclophosphamide or radiation therapy, although able to kill cells in cycle to a greater degree than cells not in cycle, can also kill nondividing cells.
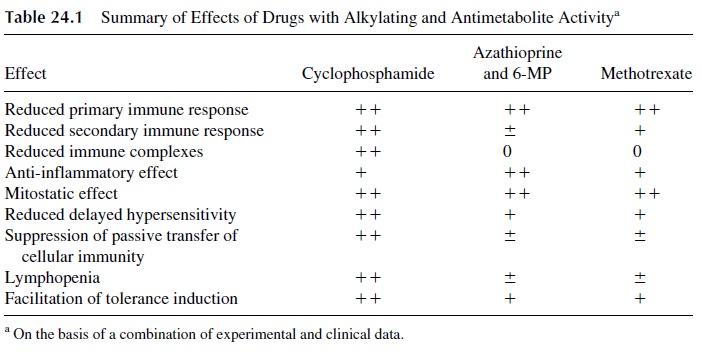
The three classical cytotoxic drugs, cyclophosphamide, azathioprine and methotrex-ate, all suppress primary and secondary humoral immune responses, delayed hypersensi-tivity, skin graft rejection, and autoimmune disease in animals. However, some striking dif-ferences in the mechanism of action of these three agents have become apparent (Table 24.1).
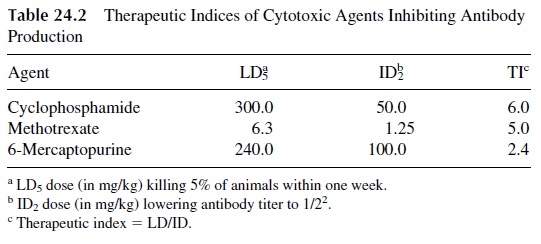
In studies of the effects of cyclophosphamide, methotrexate, and 6-MP on antibody production in mice, one can compare the dose that kills 5% of the animals within one week (LD5) with the dose required to reduce the antibody response of the mice by a factor of 2 (inhibitory dose; ID2); a therapeutic index (TI) can be calculated, which is defined as the ratio of the two doses (LD5 /ID2). Cyclophosphamide has the highest therapeutic index, fol-lowed by methotrexate and 6-MP (Table 24.2).
Sharply different effects of azathioprine and cyclophosphamide on humoral antibody production have also been demonstrated, using flagellin as a test antigen. There was a sig-nificant suppression of antibody response to flagellin in cyclophosphamide-treated pa-tients, while the responses of azathioprine-treated patients did not differ significantly from those of nontreated control patients. Several investigators have also shown that cyclophos-phamide can decrease the production of anti-DNA antibodies in both NZB mice and hu-mans. This suggests that cyclophosphamide can inhibit an ongoing immune response, whereas azathioprine and 6-MP cannot. This, of course, is the situation that one faces in the treatment of patients with autoimmune disease, since the relevant immune responses are al-ready established by the time that they are recognized and treated. In patients with systemic lupus erythematosus (SLE), cyclophosphamide treatment can reverse the deposition of im-mune complexes in the dermo-epidermal junction (which correlates with renal disease), whereas steroid therapy alone does not.
Studies of the effects of these drugs on cellular immunity have shown that all three depress cellular immunity. However, comparative studies show a greater effect with cy-clophosphamide. While both cyclophosphamide and methotrexate are more effective than 6-MP in suppressing a PPD skin test in experimental animals, only cyclophosphamide de-pletes thymus-dependent areas of the lymph nodes. The in vitro response of lymphocytes to PHA and other mitogens is likewise inhibited only by cyclophosphamide. In addition, tolerance induction is much easier to achieve in mice treated with cyclophosphamide than with azathioprine or methotrexate.
3. Inhibitors of Signal Transduction: Cyclosporine A, Tacrolimus, and Sirolimus
These compounds are fungal metabolites with immunosuppressive properties. Structurally they are macrocyclic compounds; cyclosporin A (CsA) is a macrocyclic peptide, while Tacrolimus and Sirolimus are macrocyclic lactones (macrolides). All are virtually devoid of toxicity for leukocyte precursors and, hence, do not cause leukopenia or lymphopenia. Their molecular mechanism of action depends on their binding to cytoplasmic proteins in-volved in the process of signal transduction essential for lymphocyte activation and/or pro-liferation (Fig. 24.2). Because of their ability to bind immunosuppressive compounds, these proteins are collectively known as immunophilins.
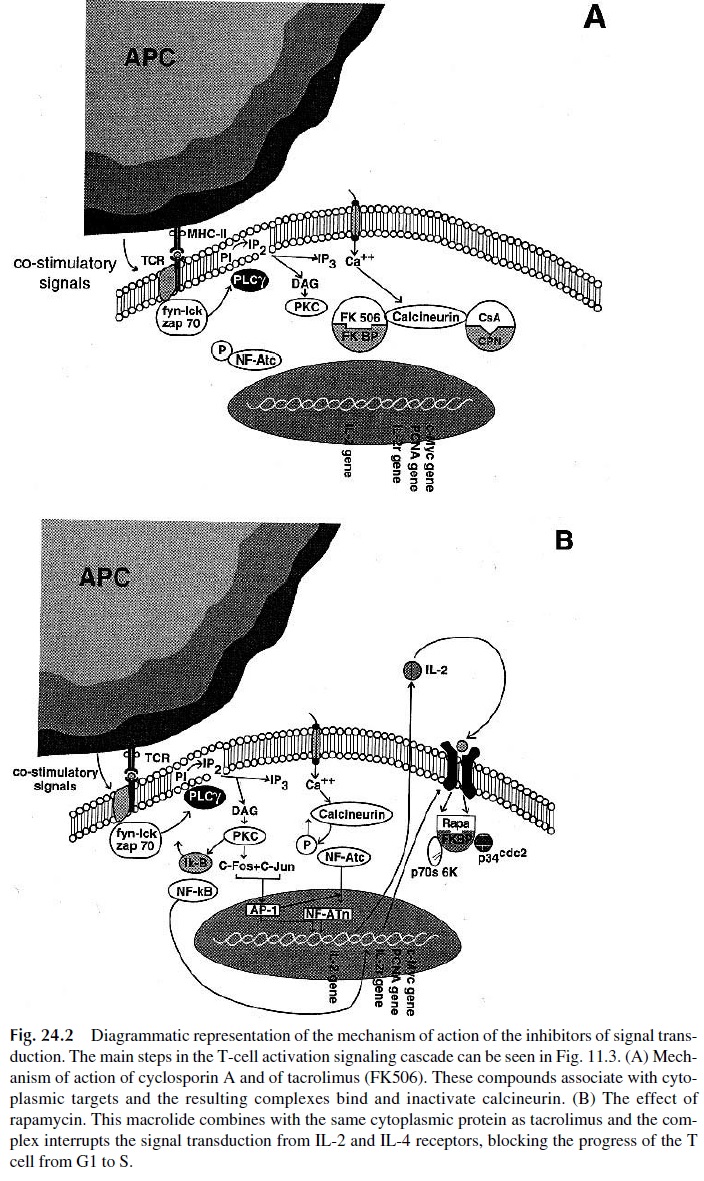
Cyclosporine A is cyclic undecapeptide obtained from Tolypocladium inflatum. It has a uniquely selective effect on T lymphocytes, suppressing humoral T-dependent re-sponses and cell-mediated immune responses. Its mechanism of action involves high-affin-ity binding to cyclophilin. The CsA-cyclophilin complex, in turn, binds and inactivates cal-cineurin. This protein, upon activation, acquires phosphatase proteins and activates NF-AT, a nuclear binding protein involved in the control of the expression of the IL-2 gene and other cytokine genes by dephosphorylation. As a consequence of the inactivation of calcineurin, Ca2+ -associated T-cell activation pathways, such as those trig-gered by anti-CD3 antibodies or the occupancy of the TcR, are inhibited and there is a gen-eral downregulation of the production of IL-2, interferon-γ , IL-3, IL-4, GM-CSF, and TNF. The expression of the CD40 ligand is also downregulated.
Helper (CD4+ ) T lymphocytes are the chief cellular target for CsA; T cells with sup-pressor activity, on the other hand, appear to proliferate at higher rates. This differential ef-fect is reflected in humans by a reversal of the CD4/CD8 ratio rates and by a relative in-crease in suppressor lymphocyte function. However, the activation of cytotoxic T lymphocytes is also inhibited, apparently due to both the lack of stimulatory signals provided by IL-2 and interferon-γ and to a direct inhibitory effect on cytotoxic T-cell precur-sors.
CsA has a remarkable ability to prolong graft survival. In experimental animals, even short courses of CsA can result in significant prolongation of kidney graft survival, sug-gesting that the drug facilitates the development of low dose tolerance. In humans, used in conjunction with azathioprine or mycophenolate and corticosteroids, it reduces the number of rejection episodes in renal transplantation, even in patients with cytotoxic antibodies and receiving poorly matched organs. It also induces a substantially longer survival of kidney, liver, and especially heart transplants, and reduces the incidence and severity of graft-ver-sus-host disease in bone marrow transplantation. Recent observations suggest that the long-term immunosuppression achieved with associations of low-dose CsA, steroids, and aza-thioprine or mycophenolate may be preferable to long-term administration of high doses of CsA, at least in patients with kidney transplants.
The main advantages of CsA as an immunosuppressant are its selective effect on T lymphocytes and its excellent steroid-sparing effect. The dosages of steroids necessary to achieve effective immunosuppression are considerably lower when steroids are used with CsA than when they are associated to other immunosuppressive drugs. As a result, the inci-dence of infection is substantially reduced, although cytomegalovirus infections are rela-tively common in CsA-treated patients. On the other hand, CsA has many serious side ef-fects, including nephrotoxicity, a major concern in patients with kidney grafts. The renal toxicity is frequently associated with hypertension, which in turn has a negative impact in all patients, especially in those receiving a heart transplant. Other side effects include hyperc-holesterolemia, hypertriglyceridemia, hirsutism, headaches, fine tremors, gingival hyperpla-sia, pancreatitis, and electrolyte abnormalities. Accelerated atherosclerosis, probably sec-ondary to therapy-related hypercholesterolemia and hypertriglyceridemia, has been observed in heart transplant recipients surviving for over 2 years, but the mechanism responsible for this complication is not clear. Finally, after long-term administration of CsA, by itself or in combination with other immunosuppressive agents (e.g., antilymphocyte globulin), there is an increased incidence of lymphoproliferative syndromes, most of them of B-cell lineage, probably resulting from the uncontrolled proliferation of the Epstein-Barr virus.
Tacrolimus (Prograf, FK506) is produced by a different fungus (Streptomycestsukubaensis). Its mechanism of action is similar to that of CsA but is 10 –100 times moreactive. The cytoplasmic target of tacrolimus is a different protein, known as FKBP (FK506 binding protein). The tacrolimus-FKBP complex has an effect very similar to that of the cy-clophilin-CsA complex—inhibiting calcineurin and preventing the activation of NF-AT. Not surprisingly, the cellular effects of FK506 and CsA are almost identical.
Clinically, tacrolimus has been mainly used to date in liver transplantation. It was first used to reverse rejection in patients unresponsive to other immunosuppressive agents. Its later use as primary immunosuppressant followed and resulted in improved patient sur-vival. Its side effects are similar to those of CsA. Neurotoxicity (including rare cases of se-vere irreversible encephalopathy), gastrointestinal intolerance, and infections (particularly by cytomegalovirus) are the most prominent complications of its use.
Sirolimus (Rapamycin, Rapamune), from Streptomyces hygroscopicus, is struc-turally similar to tacrolimus but has a different intracellular target and different pharmaco-logical properties. Sirolimus binds two proteins: FKBP, the same cyclophilin bound by tacrolimus, and the FKBP-sirolimus–associated protein (FRAP). The complex formed by cyclophilin, FKBP, and FRAP does not interact with calcineurin, but rather with other cel-lular targets, which are inactivated. The best characterized targets are two kinases, p70 ri-bosomal protein S6 kinase and p34cdc2. These enzymes appear to be activated by the inter-action of IL-2 and IL-4 with their respective receptors and are required for cells to progress through the replication cycle.
While CsA and FK506 inhibit the transition of lymphocytes from G0 to G1, sirolimus inhibits cell division later in G1, prior to entry into the S-phase. Also, sirolimus inhibits both Ca2+ -dependent and -independent activation pathways, does not inhibit IL-2 synthesis, but inhibits the response of IL-2– and IL-4–sensitive cell lines to exogenous IL-2 or IL-4. Be-cause of the different mechanism of action, sirolimus is effective even when added 12 hours after in vitro mitogenic stimulation of T cells, while CsA and FK506 are only effective when added to the cultures no later than 3 hours after the mitogen.
In addition to its FDA-approved indication in combination with cyclosporine and prednisone for the prevention of organ rejection after a kidney transplant, sirolimus is still under active investigation in solid organ transplants.
4. Blockers of MHC-II Expression
Two groups of drugs have received considerable attention for their previously unsuspected immunosuppressive and anti-inflammatory properties:
1. The antimalarial drugs chloroquine and hydroxychloroquine are widely used in the treatment of rheumatoid arthritis and other inflammatory diseases, and are also be-ing evaluated for the treatment of graft-vs.-host disease. These drugs interfere with normal lysosomal functions, increasing the lysosomal pH, inhibiting several steps in the antigen-processing reaction and thus reducing the expression of MHC-II— peptide complexes. This in turn decreases CD4 T-cell activation and downregu-lates the immune response. These drugs also appear to decrease the synthesis of pro-inflammatory cytokines by mononuclear leukocytes and promote lymphocyte apoptosis.
2. Statins, such as lovastatin, pravastatin, and atorvastatin, are HMG-CoA reductase inhibitors widely used as lipid-lowering agents. These drugs (especial-ly atorvastatin) inhibit the synthesis of the transactivational promoted for MHC-II, so that the increase in expression of MHC-II molecules seen after stimulation with interferon-γ is completely inhibited. Besides the potential-ly useful application in atherosclerosis, in which both the lipid-lowering and immunosuppressive/anti-inflammatory effects are beneficial, the statins are being evaluated for their potential benefits in rheumatoid arthritis and multiple sclerosis.
5. Immunosuppressive Polyclonal Antibody Preparations
Polyclonal antibody preparations are commonly used to treat corticosteroid-resistant acute rejection episodes in solid organ transplant recipients. Currently, there are two prepara-tions commercially available: antithymocyte globulin (equine antiserum) and thymo-globulin (rabbit antiserum) (Table 24.3). Both antisera are prepared by injecting human T lymphocytes into either a horse or a rabbit. The serum from these animals is then harvested and sterilized prior to administration into patients. Unfortunately, these preparations con-tain not only antibodies directed against T lymphocytes but also against neutrophils, ery-throcytes, and platelets. Therefore, patients treated with antilymphocyte globulin or with thymoglobulin may experience clinically significant neutropenia, anemia, and thrombo-cytopenia.
In addition, these preparations may cause serum sickness, because even in immunosuppressed patients heterologous proteins are so immunogenically potent that they end up eliciting a humoral immune response.
6. Immunosuppressive Monoclonal Antibodies and Other Biological Response Modifiers
Several types of monoclonal antibodies have been used with the purpose of suppressing the immune response. Two basic types of mechanisms of action seem to be involved: blocking of co-stimulatory signals and delivery of downregulating signals. While mur-ine monoclonal antibodies trigger immune responses that result in loss of efficiency and, in some cases, clinical manifestations of serum sickness, humanized (chimeric) monoclonal antibodies (hybrid molecules containing the variable regions of a murine monoclonal antibody and the Fc regions of a human immunoglobulin) are considerably less immunogenic.
Monoclonal Antibodies That Block Co-stimulatory Signals. Anti-CD2 mono-clonal antibodies have been shown to induce prolonged graft survival in experimental ani-mals. Its mechanism of action was initially thought to involve blocking of the CD2/LFA-3 interaction. However, it is also possible that these monoclonal antibodies deliver downreg-ulating signals to T cells.
Soluble CTLA-4, CTLA4-Ig, anti-CD80/86 monoclonal antibodies, and anti-CD40L have been shown to have immunosuppressive properties, particularly in animal models of autoimmune diseases. The immune suppressive effect is secondary to the blockage of the activation signals mediated by the CD80/86-CTLA-4 interaction or by the CD40-CD40L(CD 154) interaction. CTLA-4 Ig is a fusion protein containing the extracellular do-main of CTLA-4 and the Fc region of a human immunoglobulin. It has a longer half-life and is more effective than CTLA-4. Its mechanism of action involves inhibition of IL2-Rgene expression and block of cell division, causing T cells to remain at G0. A therapeutic trial of CTLA-4 Ig in patients with psoriasis resulted in clinical improvement in 60–80% of the subjects.
Monoclonal Antibodies That Deliver Inhibitory Signals.Several different mon-oclonal antibodies against T-cell markers (particularly anti-CD3, muromonab CD3) (Table 24.3) have been successfully used to treat acute graft rejection episodes . Muromonab CD3 appears to promote the destruction of T lymphocytes by ADCC. Also, the binding of muromonab CD3 to the CD3 protein on T lymphocytes causes the internal-ization of the complex and anergy of the T lymphocyte (Fig. 24.3).
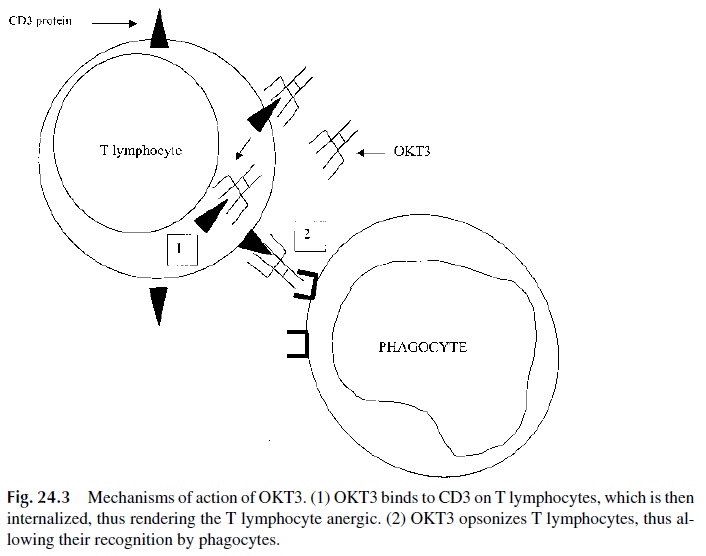
The major limitation in the clinical use of muromonab CD3 is development of human antimurine antibodies. Clinically significant antibody titers to murine immunoglobulins occur in >50% of patients treated with a full course of muromonab CD3.
Monoclonal Antibodies That Block the Interleukin-2 Receptor. Daclizumab(Zenapax) and basiliximab (Simulect) are chimeric monoclonal antibodies directed against the IL-2 receptor. Chimeric antibodies consist of murine CDR regions of an antibody in-corporated into a human IgG molecule. The alpha chain of the IL-2 receptor is an ideal can-didate for immunosuppression because it is minimally expressed on resting T cells and sig-nificantly upregulated in activated T lymphocytes. Therefore, basiliximab and daclizumab target mostly activated T lymphocytes. The proposed mechanism of action is the saturation of IL-2 receptors, thereby inhibiting IL-2–driven proliferation, but this may prove to be an oversimplistic explanation. Both basiliximab and daclizumab are used as prophylaxis for acute rejection in renal transplantation but are being investigated in other organ types (e.g., liver, bone marrow) and for reversal of acute rejection episodes.
Related Topics