Chapter: Medical Physiology: Red Blood Cells, Anemia, and Polycythemia
Formation of Hemoglobin
Formation of Hemoglobin
Synthesis of hemoglobin begins in the proerythro-blasts and continues even into the reticulocyte stage of the red blood cells. Therefore, when reticulocytes leave the bone marrow and pass into the blood stream, they continue to form minute quantities of hemoglobin for another day or so until they become mature erythro-cytes.
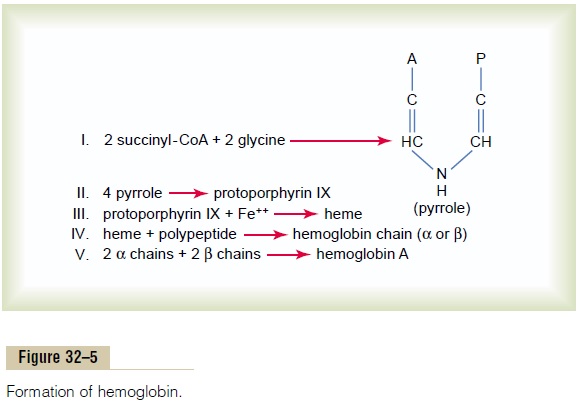
Figure 32–5 shows the basic chemical steps in the formation of hemoglobin. First, succinyl-CoA, formed in the Krebs metabolic cycle, binds with glycine to form a pyrrole molecule. In turn, four pyrroles combine to form protoporphyrin IX, which then combines with iron to form the heme molecule. Finally, each heme molecule combines with a long polypeptide chain, a globin synthesized by ribosomes, forming a subunit of hemoglobin called a hemoglobin chain (Figure 32–6). Each chain has amolecular weight of about 16,000; four of these in turn bind together loosely to form the whole hemoglobin molecule.
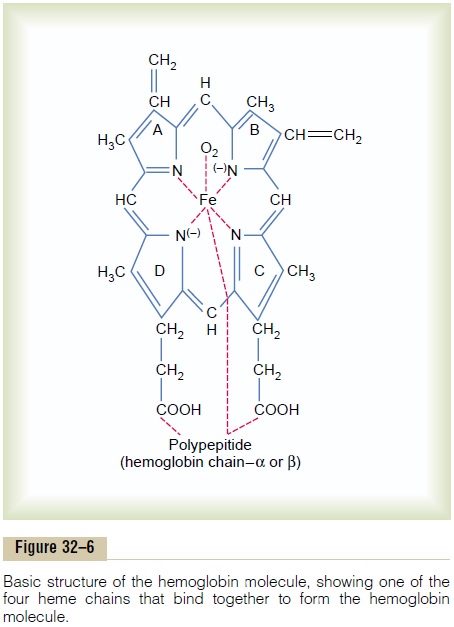
There are several slight variations in the different subunit hemoglobin chains, depending on the amino acid composition of the polypeptide portion. The dif-ferent types of chains are designated alpha chains, betachains, gamma chains, and delta chains. The mostcommon form of hemoglobin in the adult human being, hemoglobin A, is a combination of two alphachains and two beta chains.Hemoglobin A has amolecular weight of 64,458.
Because each hemoglobin chain has a heme pros-thetic group containing an atom of iron, and because there are four hemoglobin chains in each hemoglobin molecule, one finds four iron atoms in each hemoglo-bin molecule; each of these can bind loosely with one molecule of oxygen, making a total of four molecules of oxygen (or eight oxygen atoms) that can be trans-ported by each hemoglobin molecule.
The types of hemoglobin chains in the hemoglobin molecule determine the binding affinity of the hemo-globin for oxygen. Abnormalities of the chains can alter the physical characteristics of the hemoglobin molecule as well. For instance, in sickle cell anemia, the amino acid valine is substituted for glutamic acid at one point in each of the two beta chains.
When this type of hemoglobin is exposed to low oxygen, it forms elongated crystals inside the red blood cells that are sometimes 15 micrometers in length. These make it almost impossible for the cells to pass through many small capillaries, and the spiked ends of the crystals are likely to rupture the cell membranes, leading to sickle cell anemia.
Combination of Hemoglobin with Oxygen. The most important feature of the hemoglobin molecule is its ability to combine loosely and reversibly with oxygen. This ability is discussed in detail in relation to respiration, because the primary function of hemo-globin in the body is to combine with oxygen in the lungs and then to release this oxygen readily in the peripheral tissue capillaries, where the gaseous tension of oxygen is much lower than in the lungs.
Oxygen does not combine with the two positive bonds of the iron in the hemoglobin molecule. Instead, it binds loosely with one of the so-called coordination bonds of the iron atom. This is an extremely loose bond, so that the combination is easily reversible. Fur-thermore, the oxygen does not become ionic oxygen but is carried as molecular oxygen (composed of two oxygen atoms) to the tissues, where, because of the loose, readily reversible combination, it is released into the tissue fluids still in the form of molecular oxygen rather than ionic oxygen.
Related Topics