Chapter: Basic & Clinical Pharmacology : Special Aspects of Perinatal & Pediatric Pharmacology
Drug Therapy in Infants & Children
DRUG THERAPY IN INFANTS &
CHILDREN
Physiologic
processes that influence pharmacokinetic variables in the infant change
significantly in the first year of life, particularly during the first few
months. Therefore, special attention must be paid to pharmacokinetics in this
age group. Pharmacodynamic differences between pediatric and other patients
have not been explored in great detail and are probably small except for those
specific target tissues that mature at birth or immediately thereaf-ter (eg,
the ductus arteriosus).
Drug Absorption
Drug
absorption in infants and children follows the same general principles as in
adults. Unique factors that influence drug absorp-tion include blood flow at
the site of administration, as deter-mined by the physiologic status of the
infant or child; and, for orally administered drugs, gastrointestinal function,
which changes rapidly during the first few days after birth. Age after birth
also influences the regulation of drug absorption.
A. Blood Flow at the Site of Administration
Absorption after
intramuscular or subcutaneous injection depends mainly, in neonates as in
adults, on the rate of blood flow to the muscle or subcutaneous area injected.
Physiologic conditions that might reduce blood flow to these areas are
cardiovascular shock, vasoconstriction due to sympathomimetic agents, and heart
failure. However, sick preterm infants requiring intramuscular injections may
have very little muscle mass. This is further complicated by diminished
peripheral perfusion to these areas. In such cases, absorption becomes
irregular and difficult to predict, because the drug may remain in the muscle
and be absorbed more slowly than expected. If perfusion suddenly improves,
there can be a sudden and unpredictable increase in the amount of drug entering
the circula-tion, resulting in high and potentially toxic concentrations of
drug. Examples of drugs especially hazardous in such situations are cardiac
glycosides, aminoglycoside antibiotics, and anticonvulsants.
B. Gastrointestinal Function
Significant
biochemical and physiologic changes occur in the neo-natal gastrointestinal
tract shortly after birth. In full-term infants, gastric acid secretion begins
soon after birth and increases gradu-ally over several hours. In preterm
infants, the secretion of gastric acid occurs more slowly, with the highest
concentrations appear-ing on the fourth day of life. Therefore, drugs that are
usually partially or totally inactivated by the low pH of gastric contents
should not be administered orally.
Gastric emptying time
is prolonged (up to 6 or 8 hours) in the first day or so after delivery.
Therefore, drugs that are absorbed primarily in the stomach may be absorbed
more completely than anticipated. In the case of drugs absorbed in the small
intestine, therapeutic effect may be delayed. Peristalsis in the neonate is
irregular and may be slow. The amount of drug absorbed in the small intestine
may therefore be unpredictable; more than the usual amount of drug may be
absorbed if peristalsis is slowed, and this could result in potential toxicity
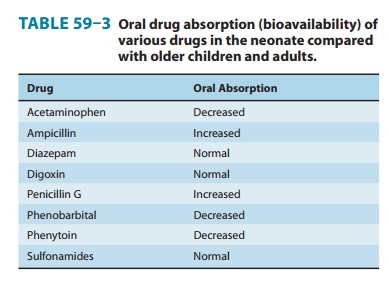
from an otherwise standard dose. Table 59–3 summarizes data on oral
bioavailability of various drugs in neonates compared with older
children and adults. An increase in peristalsis, as in diarrheal conditions,
tends to decrease the extent of absorption, because contact time with the large
absorptive surface of the intestine is decreased.
Gastrointestinal
enzyme activities tend to be lower in the new-born than in the adult.
Activities of α-amylase
and other pancre-atic enzymes in the duodenum are low in infants up to 4 months
of age. Neonates also have low concentrations of bile acids and lipase, which
may decrease the absorption of lipid-soluble drugs.
Drug Distribution
As body composition
changes with development, the distribution volumes of drugs are also changed.
The neonate has a higher per-centage of its body weight in the form of water
(70–75%) than does the adult (50–60%). Differences can also be observed between
the full-term neonate (70% of body weight as water) and the small preterm
neonate (85% of body weight as water). Similarly, extracellular water is 40% of
body weight in the neo-nate, compared with 20% in the adult. Most neonates will
experi-ence diuresis in the first 24–48 hours of life. Since many drugs are
distributed throughout the extracellular water space, the size (vol-ume) of the
extracellular water compartment may be important in determining the
concentration of drug at receptor sites. This is especially important for
water-soluble drugs (such as aminoglyco-sides) and less crucial for
lipid-soluble agents.Preterm infants have much less fat
than full-term infants. Total body fat in preterm infants is about 1% of total
body weight, compared with 15% in full-term neonates. Therefore, organs that
generally accumulate high concentrations of lipid-soluble drugs in adults and
older children may accumulate smaller amounts of these agents in less mature
infants.
Another major factor
determining drug distribution is drug binding to plasma proteins. Albumin is
the plasma protein with the greatest binding capacity. In general, protein
binding of drugs is reduced in the neonate. This has been seen with local
anesthetic drugs, diazepam, phenytoin, ampicillin, and phenobarbital.
Therefore, the concentration of free (unbound) drug in plasma is
Because the free drug exerts the pharmacologic effect, this can result in
greater drug effect or toxicity despite a normal or even low plasma concentration
of total drug (bound plus unbound). Consider a therapeutic dose of a drug (eg,
diaze-pam) given to a patient. The concentration of total drug in the plasma is
300 mcg/L. If the drug is 98% protein-bound in an older child or adult, then 6
mcg/L is the concentration of free drug. Assume that this concentration of free
drug produces the desired effect in the patient without producing toxicity.
However, if this drug is given to a preterm infant in a dosage adjusted for
body weight and it produces a total drug concentration of 300 mcg/L—and protein
binding is only 90%—then the free drug concentration will be 30 mcg/L, or five
times higher. Although the higher free concentration may result in faster
elimination , this concentration may be quite toxic initially.
Some
drugs compete with serum bilirubin for binding to albu-min. Drugs given to a
neonate with jaundice can displace biliru-bin from albumin. Because of the
greater permeability of the neonatal blood-brain barrier, substantial amounts
of bilirubin may enter the brain and cause kernicterus. This was in fact
observed when sulfonamide antibiotics were given to preterm neonates as
prophylaxis against sepsis. Conversely, as the serum bilirubin rises for
physiologic reasons or because of a blood group incompatibil-ity, bilirubin can
displace a drug from albumin and substantially raise the free drug
concentration. This may occur without altering the total drug concentration and
would result in greater therapeu-tic effect or toxicity at normal
concentrations. This has been shown to happen with phenytoin.
Drug Metabolism
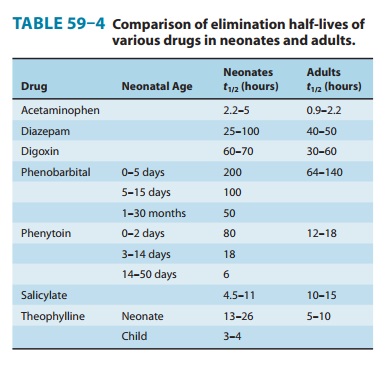
The metabolism of most drugs occurs in the liver . The drug-metabolizing activities of the cytochrome P450-dependent mixed-function oxidases and the conjugating enzymes are substantially lower (50–70% of adult values) in early neonatal life than later. The point in development at which enzymatic activity is maximal depends upon the specific enzyme system in question. Glucuronide formation reaches adult values (per kilo-gram body weight) between the third and fourth years of life. Because of the neonate’s decreased ability to metabolize drugs, many drugs have slow clearance rates and prolonged elimination half-lives. If drug doses and dosing schedules are not altered appropriately, this immaturity predisposes the neonate to adverse effects from drugs that are metabolized by the liver. Table 59–4 demonstrates how neonatal and adult drug elimination half-lives can differ and how the half-lives of phenobarbital and phenytoin decrease as the neonate grows older. The process of maturation must be considered when administering drugs to this age group, especially in the case of drugs administered over long periods.Another consideration for the neonate is whether or not the mother was receiving drugs (eg, phenobarbital) that can induce early maturation of fetal hepatic enzymes. In this case, the ability of the neonate to metabolize certain drugs will be greater than expected, and one may see less therapeutic effect and lower plasma drug concentrations when the usual neonatal dose is given. During toddlerhood (12–36 months), the metabolic rate of many drugs exceeds adult values, often necessitating larger doses per kilogram than later in life.
Drug Excretion
The glomerular
filtration rate is much lower in newborns than in older infants, children, or
adults, and this limitation persists during the first few days of life.
Calculated on the basis of body surface area, glomerular filtration in the
neonate is only 30–40% of the adult value. The glomerular filtration rate is
even lower in neonates born before 34 weeks of gestation. Function improves
substantially during the first week of life. At the end of the first week, the
glom-erular filtration rate and renal plasma flow have increased 50% from the
first day. By the end of the third week, glomerular filtra-tion is 50–60% of
the adult value; by 6–12 months, it reaches adult values (per unit surface
area). Therefore, drugs that depend on renal function for elimination are
cleared from the body very slowly in the first weeks of life. Subsequently,
during toddlerhood, it exceeds adult values, often necessitating larger doses
per kilogram than in adults, as described previously for drug-metabolic rate.
Penicillins, for example, are cleared by preterm infants at 17% of the adult rate based on comparable surface area and 34% of the adult rate when adjusted for body weight. The dosage of ampicillin for a neonate less than 7 days old is 50–100 mg/kg/d in two doses at 12-hour intervals. The dosage for a neonate over 7 days old is 100–200 mg/kg/d in three doses at 8-hour intervals. A decreased rate of renal elimination in the neonate has also been observed with aminoglycoside antibiotics (kanamycin, gentamicin, neomycin, and streptomycin). The dosage of gentamicin for a neonate less than 7 days old is 5 mg/kg/d in two doses at 12-hour intervals. The dosage for a neonate over 7 days old is 7.5 mg/kg/d in three doses at 8-hour intervals. Total body clearance of digoxin is directly dependent upon adequate renal function, and accumulation of digoxin can occur when glomerular filtration is decreased. Since renal function in a sick infant may not improve at the predicted rate during the first weeks and months of life, appropriate adjustments in dosage and dosing schedules may be very difficult. In this situation, adjust-ments are best made on the basis of plasma drug concentrations determined at intervals throughout the course of therapy.
Although great focus
is naturally concentrated on the neonate, it is important to remember that
toddlers may have shorter
elimi-nation half-lives of drugs than older children and adults, due probably
to increased renal elimination and
metabolism. For example, the dose per kilogram of digoxin is much higher in
tod-dlers than in adults. The mechanisms for these developmental changes are
still poorly understood.
Special Pharmacodynamic Features in the Neonate
The
appropriate use of drugs has made possible the survival of neo-nates with
severe abnormalities who would otherwise die within days or weeks after birth.
For example, administration of indomethacin
causes the rapid closure of a patent ductus arterio-sus, which would
otherwise require surgical closure in an infant with a normal heart. Infusion
of prostaglandin E1, on the
other hand, causes the ductus to remain open, which can be lifesaving in an
infant with transposition of the great vessels or tetralogy of Fallot . An
unexpected effect of such infusion has been described. The drug caused antral
hyperplasia with gastric outlet obstruction as a clinical manifestation in 6 of
74 infants who received it. This phenomenon appears to be dose-dependent.
Neonates are also more sensitive to the central depressant effects of opioids
than are older children and adults, necessitating extra caution when they are
exposed to some narcotics (eg, codeine) through breast milk.
Related Topics