Chapter: Clinical Cases in Anesthesia : Cardiomyopathy Managed With A Left Ventricular Assist Device
Distinguish dilated cardiomyopathy from other cardiomyopathies
Distinguish
dilated cardiomyopathy from other cardiomyopathies.
Dilated cardiomyopathy (DCM) is characterized by impaired systolic function and dilation of the left ventricle (LV) or sometimes both ventricles. The ejection fraction is moderately-to-severely depressed, and both tricuspid and mitral regurgitation generally result from stretching of their respective annuli. Diastolic dysfunction typically develops as the disease progresses, and pulmonary artery (PA) pressures are elevated secondary to chronically high left atrial (LA) pressures. Of the three predominant primary cardiomyopathies (dilated, restrictive, and hyper-trophic), DCM is the most common. DCM may result from infectious entities, cytotoxic substances, metabolic errors, endocrinopathies, familial inheritance, or immuno-logic mechanisms. Where no specific etiology can be identified, the term idiopathic dilated cardiomyopathy is applied. An excellent review of DCMs was recently published by Felker et al. (1999) (Table 11.2). It should be understood, however, that dilation of the heart chambers is a final, common outcome of many pathophysiologic processes, not all of which represent primary DCM. While a dilated cardiomyopathy is not always DCM, per se, the anesthetic considerations and management are the same.
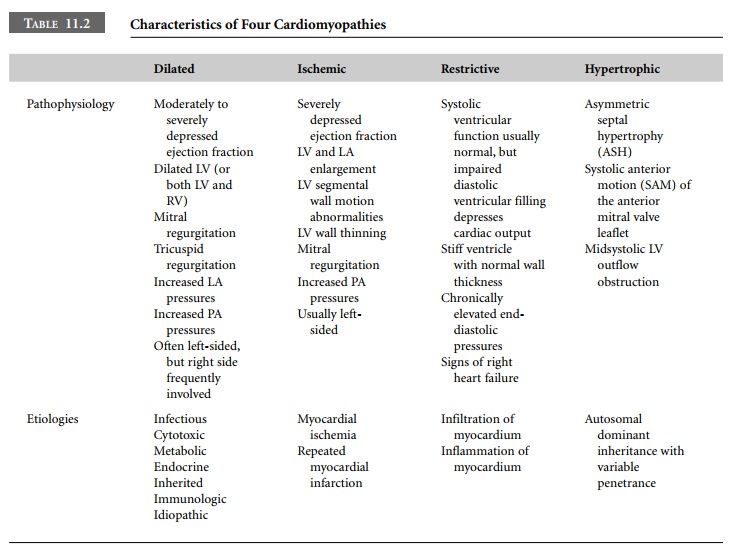
Severe LV dysfunction resulting from recurrent myocar-dial infarctions is commonly termed ischemic cardiomy-opathy (ICM). ICM is not a primary cardiomyopathy because the primary underlying etiology is coronary artery disease. ICM presents with a similar pattern of impairment as DCM. ICM is characterized by LA and LV enlargement, LV segmental wall motion abnormalities, LV wall thinning, severely depressed ejection fraction, mitral regurgitation due to stretching of the mitral annulus, and elevated pulmonary artery pressures. Diastolic dysfunction is often present. In the absence of right ventricular (RV) infarction, RV size and function can be normal. While ICM frequently causes cardiac chamber dilation, a history of repeated myocardial infarctions and predominantly left-sided dilation and dysfunction helps to distinguish ICM from DCM.
Restrictive cardiomyopathy (RCM) usually
results from infiltration or inflammation of the myocardium. The result is a
stiff ventricle with an abnormally restrictive pattern of diastolic filling,
and chronically elevated end-diastolic pressures are characteristic of this
disease. While systolic ventricular function and wall thickness are usually
normal, impairment of diastolic filling results in depressed cardiac output.
Many patients with RCM present with signs of right-sided heart failure
(peripheral edema, jugular venous distention, ascites, and hepatomegaly without
shortness of breath or pulmonary rales). True RCM is relatively uncommon.
Hypertrophic cardiomyopathy (HCM) generally devel-ops
from asymmetric hypertrophy of the ventricular septum. Some patients with HCM
have an obstructive form, hypertrophic obstructive cardiomyopathy (HOCM). In
HOCM, systolic anterior motion (SAM) of the anterior mitral valve leaflet
contacts the hypertrophied basal septum, producing mid-systolic obstruction of
the left ventricular outflow tract. Those afflicted with HOCM are at risk for
sudden death. Nonobstructive forms of hyper-trophic cardiomyopathy also exist.
HCM is an autosomal dominant disease with variable penetrance. Long-standing
hypertension can also produce significant LV hypertrophy (often termed
hypertensive heart disease, HHD). While diastolic dysfunction due to
hypertrophied myocardium, and therefore some degree of pulmonary hypertension, tend
to be present in both diseases, their characteristics and management are
different. LV hypertrophy is asymmetric in HCM, but is concentric in HHD.
Systolic function in HCM tends to be normal, while cardiac output tends to
decline as HHD progresses. Systolic obstruction to LV outflow is a dynamic,
subaortic process in HOCM and in HHD there is midventricular cavitary
obliteration.
Related Topics