Chapter: Obstetrics and Gynecology: Puberty
Delayed Puberty
Delayed Puberty
There is wide variation in normal
pubertal development. However, puberty is
considered delayed when secondary sex characteristics have not appeared by age
13, there is no evidence of menarche by age 15 to 16, or when menses have not
began 5 years after the onset of thelarche. These findings shouldprompt the
physician to initiate a workup to determine the cause of the delay. The most
common causes of delayed puberty are shown in Box 34.2.
HYPERGONADOTROPIC HYPOGONADISM
The most
common cause of delayed puberty with an elevated FSH is gonadal dysgenesis, or
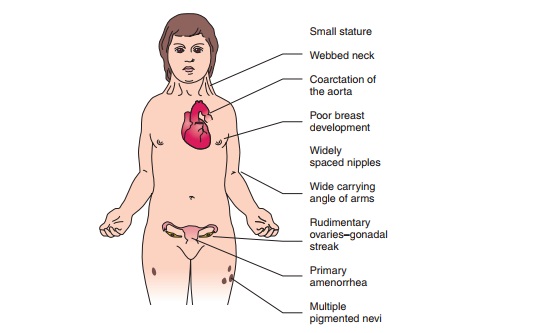
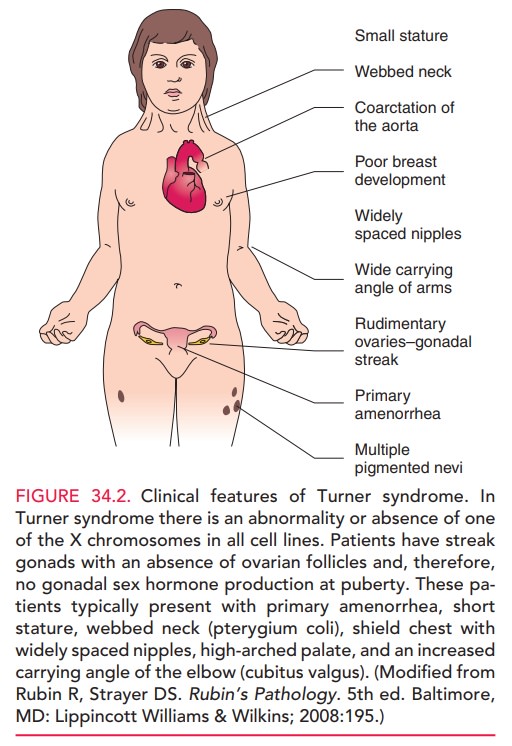
Turner syndrome. In this condition,there is an
abnormality in or absence of one of the X chro-mosomes in all cell lines.
Patients have streak gonads, with an absence of ovarian follicles; therefore,
gonadal sex hor-mone production does not occur at puberty. These pa-tients
typically have primary amenorrhea, short stature, webbed neck (pterygium coli),
shield chest with widely spaced nipples, high arched palate, and an increased
carry-ing angle of the elbow (cubitus valgus) [see Fig. 34.2].
Estrogen
administration should be initiated at the normal time of initiation of puberty,
and growth hormone should be initiated very early (often prior to estrogen
therapy) and ag-gressively to normalize adult height.
Estrogen is necessary to
stimulate breast development, genital tract maturation, and the beginning of
menstrua-tion. Low-dose estrogen is used to initiate secondary sexual
maturation, and the dose is increased once breast budding and menarche occur.
If an excessive amount of
Box 34.2
Causes of Delayed Puberty
Hypergonadotropic Hypogonadism (FSH >30 mIU/mL)
·
Gonadal
dysgenesis (Turner syndrome)
Hypogonadotropic Hypogonadism (FSH LH <10 mIU/mL)
·
Constitutional
(physiologic) delay
·
Kallmann
syndrome
·
Anorexia/Extreme
exercise
·
Pituitary
tumors/pituitary disorders
·
Hyperprolactinemia
·
Drug
use
Anatomic Causes
·
Müllerian
agenesis
·
Imperforate
hymen
·
Transverse
vaginal septum
estrogen is administered
initially, epiphyseal closure may begin, and long bone growth is truncated and
adult height compromised. A delay in estrogen administration can lead to the
development of osteoporosis in the teenage years. Progestins should not be
given until the patient has reached Tanner stage IV, because premature
progestin therapy may prevent the breast from developing completely, thus
result-ing in an abnormal contour (a more tubular breast).
HYPOGONADOTROPIC HYPOGONADISM
The arcuate nucleus of the
hypothalamus secretes GnRH in cyclic bursts (or a pulsatile fashion), which
stimulates release of gonadotropins from the anterior pituitary gland. Dysfunction
of the arcuate nucleus disrupts the short hormonal loop between the
hypothalamus and pi-tuitary. As a result, FSH and LH secretion does not occur.
Consequently, the ovaries are not stimulated to secrete estradiol, and
secondary sexual maturation is delayed. Themost
common cause of this type of delayed puberty is constitutional (physiologic)
delay. Other causes include Kallmann syndrome;anorexia, exercise, or
stress; pituitary tumors/pituitary dis-orders; hyperprolactinemia; and drug
use.
Constitutional delay of puberty
represents approxi-mately 20% of all cases of delayed puberty. It is thought to bea normal variant of the
development process and trends can be seen within families. Children with
constitutional delay usuallyhave not only delay of secondary sexual maturation,
but also short stature with an appropriate delay of bone maturation.
In the
Kallmann syndrome, the olfactory tracts are hypoplas-tic, and the arcuate
nucleus does not secrete GnRH. Youngwomen with Kallmann syndrome
have little or no sense of smell and do not have breast development. This
condition can be diagnosed on initial physical examination by chal-lenging the
olfactory function with known odors such as coffee or rubbing alcohol. Once the
condition is recog-nized and treated, the prognosis for successful secondary
sexual maturation and reproduction is excellent. Secondary sexual maturation
can be stimulated by the administration of exogenous hormones or by the
administration of pul-satile GnRH. Patients typically can have normal reproduc-tive
function. Ovulation is induced by the administration of exogenous gonadotropin,
and progesterone is given in the luteal phase to allow implantation of the
embryo.
Other
causes of hypothalamic amenorrhea include weight loss, strenuous exercise (such
as ballet dancing or long-distance running), anorexia nervosa, or bulimia. These
conditions allresult in suppressed gonadotropin levels with low estrogen
levels. The correction of the underlying abnormality (such as weight gain in
patients with weight loss) restores normal gonadotropin levels, stimulating
ovarian steroidogenesis and the resumption of pubertal development.
Craniopharyngioma
is the most common tumor associated with delayed puberty. This
tumor develops in the pituitarystalk with suprasellar extension from nests of
epithelium de-rived from the Rathke pouch. The radiologic hallmark is the
appearance of a (supra)sellar calcified cyst. Calcifications are present in
approximately 70% of craniopharyngiomas.
ANATOMIC CAUSES
During fetal life, müllerian
ducts develop and fuse in the female fetus to form the upper reproductive tract
(i.e., the fallopian tubes, uterus, and upper vagina). The lower and midportion
of the vagina develop from the canalization of the genital plate.
Müllerian
agenesis, or Mayer-Rokitansky–Küster–Hauser syndrome, is the most common cause
of primary amenorrhea in women with normal breast development. In this
syndrome,there is congenital absence of the vagina and usually an ab-sence of
the uterus and fallopian tubes. Ovarian function is normal, because the ovaries
are not derived from müller-ian structures; therefore, all the secondary sexual
charac-teristics of puberty occur at the appropriate time. Physical examination
leads to the diagnosis of müllerian agenesis. Renal anomalies (e.g.,
reduplication of the ureters, horse-shoe kidney, or unilateral renal agenesis)
occur in 40% to 50% of cases. Skeletal anomalies such as scoliosis occur in 10%
to 15% of cases. Mayer-Rokitansky–Küster–Hauser syndrome is generally sporadic in
expression, although oc-casional occurrences in families can be seen.
There are several therapeutic
approaches to this con-dition. Nonsurgical approaches should be tried first,
using dilators and pressure on the dimple between the urethra and the rectum,
twice a day. This tissue is quite pliable and, with increasing dilator size, a
normal-length vagina can be achieved. An artificial vagina may be created by
repetitive pressure by vaginal dilators on the perineum or by surgi-cal
construction followed by a split-thickness skin graft. After creation of a
vagina, these women are able to have sexual intercourse. With the advances in
assisted repro-ductive technologies, including in vitro fertilization (IVF) and
use of a surrogate mother (gestational carrier), it is possible for a woman
with this condition to have a genetic child by using her oocytes.
The
simplest genital tract anomaly is imperforate hymen. In this
condition, the genital plate canalization is incom-plete, and the hymen is,
therefore, closed. Menarche occurs at the appropriate time, but because there
is obstruction to the passage of menstrual blood, it is not apparent. This
condition presents with pain in the area of the uterus and a bulging,
bluish-appearing vaginal introi-tus. Hymenotomy is the definitive therapy. This
condi-tion may be confused with a transverse vaginal septum. Transverse vaginal
septa can occur along the vagina at any level and result in obstruction to
outflow of menses. A vaginal septum can be resected and primarily repaired via procedure
called a Z-vaginoplasty. Prolonged obstruc-tion to menstruation can be
associated with an increased incidence of endometriosis.

Related Topics