Chapter: Basic & Clinical Pharmacology : Tetracyclines, Macrolides,Clindamycin,Chloramphenicol,Streptogramins,& Oxazolidinones
Chloramphenicol
CHLORAMPHENICOL
Crystalline
chloramphenicol is a neutral, stable compound with the following structure:
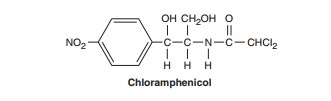
It is soluble in
alcohol but poorly soluble in water. Chlorampheni-col succinate, which is used
for parenteral administration, is highly water-soluble. It is hydrolyzed in
vivo with liberation of free chloramphenicol.
Mechanism of Action & Antimicrobial Activity
Chloramphenicol
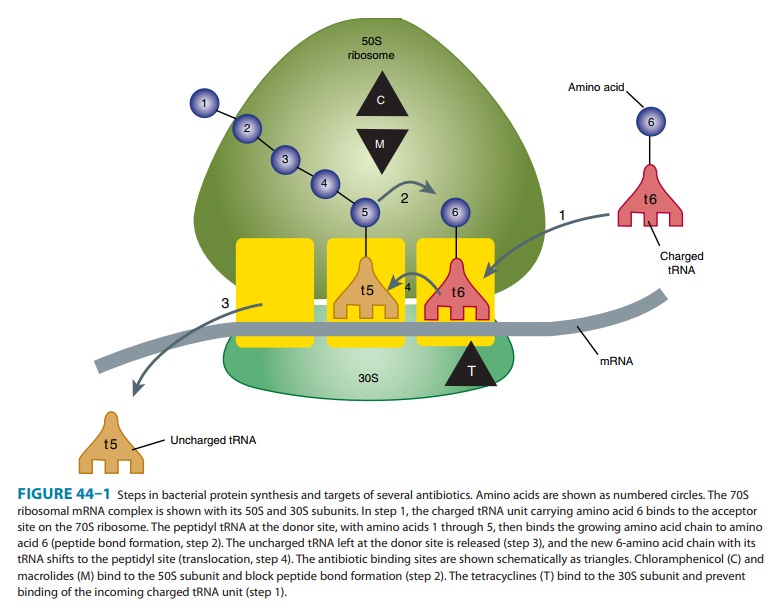
is a potent inhibitor of microbial protein synthesis. It binds reversibly to
the 50S subunit of the bacterial ribosome
(Figure
44–1) and inhibits peptide bond formation (step 2). Chloramphenicol is a
bacteriostatic broad-spectrum antibiotic that is active against both aerobic
and anaerobic gram-positive and gram-negative organisms. It is active also
against Rickettsiae but not Chlamydiae. Most gram-positive bacteria
are inhibited at concentra-tions of 1–10 mcg/mL, and many gram-negative
bacteria are inhib-ited by concentrations of 0.2–5 mcg/mL. H influenzae, Neisseriameningitidis, and some strains of
bacteroides are highly susceptible,and for these organisms, chloramphenicol may
be bactericidal.
Low-level resistance
to chloramphenicol may emerge from large populations of
chloramphenicol-susceptible cells by selec-tion of mutants that are less
permeable to the drug. Clinically significant resistance is due to production
of chloramphenicol acetyltransferase, a plasmid-encoded enzyme that inactivates
the drug.
Pharmacokinetics
The usual dosage of
chloramphenicol is 50–100 mg/kg/d. After oral administration, crystalline
chloramphenicol is rapidly and completely absorbed. A 1-g oral dose produces
blood levels between 10 and 15 mcg/mL. Chloramphenicol palmitate is a pro-drug
that is hydrolyzed in the intestine to yield free chlorampheni-col. The
parenteral formulation is a prodrug, chloramphenicol succinate, which
hydrolyzes to yield free chloramphenicol, giving blood levels somewhat lower
than those achieved with orally administered drug. Chloramphenicol is widely
distributed to virtu-ally all tissues and body fluids, including the central
nervous system and cerebrospinal fluid, such that the concentration of
chloram-phenicol in brain tissue may be equal to that in serum. The drug
penetrates cell membranes readily.
Most of the drug is
inactivated either by conjugation with glucuronic acid (principally in the
liver) or by reduction to inac-tive aryl amines. Active chloramphenicol (about
10% of the total dose administered) and its inactive degradation products
(about 90% of the total) are eliminated in the urine. A small amount of active
drug is excreted into bile and feces. The systemic dosage of chloramphenicol
need not be altered in renal insufficiency, but it must be reduced markedly in
hepatic failure. Newborns less than a week old and premature infants also clear
chloramphenicol less well, and the dosage should be reduced to 25 mg/kg/d.
Clinical Uses
Because
of potential toxicity, bacterial resistance, and the avail-ability of many
other effective alternatives, chloramphenicol is rarely used in the United
States. It may be considered for treat-ment of serious rickettsial infections
such as typhus and Rocky Mountain spotted fever. It is an alternative to a β-lactam
antibiotic for treatment of bacterial meningitis occurring in patients who have
major hypersensitivity reactions to penicillin. The dosage is 50–100 mg/kg/d in
four divided doses.
Chloramphenicol
is used topically in the treatment of eye infections because of its broad
spectrum and its penetration of ocular tissues and the aqueous humor. It is
ineffective for chla-mydial infections.
Adverse Reactions
Adults occasionally
develop gastrointestinal disturbances, includ-ing nausea, vomiting, and diarrhea.
This is rare in children. Oral or vaginal candidiasis may occur as a result of
alteration of normal microbial flora.
Chloramphenicol
commonly causes a dose-related reversible suppression of red cell production at
dosages exceeding 50 mg/ kg/d after 1–2 weeks. Aplastic anemia, a rare
consequence (1 in 24,000 to 40,000 courses of therapy) of chloramphenicol
admin-istration by any route, is an idiosyncratic reaction unrelated to dose,
although it occurs more frequently with prolonged use. It tends to be irreversible
and can be fatal.
Newborn
infants lack an effective glucuronic acid conjugation mechanism for the
degradation and detoxification of chloram-phenicol. Consequently, when infants
are given dosages above 50 mg/kg/d, the drug may accumulate, resulting in the gray babysyndrome, with vomiting,
flaccidity, hypothermia, gray color, shock,and vascular collapse. To avoid this
toxic effect, chloramphenicol should be used with caution in infants and the
dosage limited to 50 mg/kg/d (or less during the first week of life) in
full-term infants more than 1 week old and 25 mg/kg/d in premature infants.
Chloramphenicol
inhibits hepatic microsomal enzymes that metabolize several drugs. Half-lives
of these drugs are prolonged, and the serum concentrations of phenytoin,
tolbutamide, chlorpropa-mide, and warfarin are increased. Like other
bacteriostatic inhibitors of microbial protein synthesis, chloramphenicol can
antagonize bac-tericidal drugs such as penicillins or aminoglycosides.
Related Topics