Chapter: Modern Pharmacology with Clinical Applications: Introduction to Central Nervous System Pharmacology
Central Nervous System Neurotransmitters
CENTRAL NERVOUS
SYSTEM NEUROTRANSMITTERS
A large number of CNS
neurotransmitters have been either tentatively or positively identified. While
a de-tailed discussion of the various central neurotransmit-ters and the
criteria for their identification is beyond the scope of this text, a summary
of the most important mammalian central neurotransmitters follows.
Acetylcholine
The discovery that ACh was a
transmitter in the periph-eral nervous system formed the basis for the theory
of neurotransmission. ACh is also a neurotransmitter in the mammalian brain;
however, only a few cholinergic tracts have been clearly delineated.ACh is an
excitatory neurotransmitter in the mammalian CNS. There is good evidence that
ACh (among other neurotransmitters) is decreased in certain cognitive
disorders, such as Alzheimer’s disease.
Dopamine
Quantitatively, dopamine is the
most important of the biogenic amine neurotransmitters in the CNS. The three
major distinct dopaminergic systems in the mammalian brain are categorized
according to the lengths of the neurons. There is a system comprising
ultrashort neu-rons within amacrine cells of the retina and peri-glomerular
cells in the olfactory bulb. Of the several intermediate-length dopaminergic
neuronal systems, the best studied are neurons in the tuberobasal ventral
hy-pothalamus that innervate the median eminence and the intermediate lobe of
the pituitary. These neurons are important in the regulation of various
hypothalamohy-pophysial functions, including prolactin release from the
anterior pituitary. The best-categorized of the dopamine neuronal systems are
the long projections from nuclei in the substantia nigra and ventral tegmental
areas to the limbic cortex; other limbic structures, including the amygdaloid
complex and piriform cortex; and the neo-striatum (primarily the caudate and
putamen). In Parkinson’s disease, the primary biochemical feature is a marked
reduction in the concentration of dopamine in this long projection system .
Several classes of drugs,
notably the antipsychotics, interfere with dopaminergic transmission. In
general, dopamine appears to be an in-hibitory neurotransmitter. Five dopamine
receptors have been identified; the most important and best stud-ied are the D1-
and D2-receptor groups. The D1-receptor, which increases
cyclic adenosine monophosphate (cAMP) by activation of adenylyl cyclase, is
located pri-marily in the region of the putamen, nucleus accum-bens, and in the
olfactory tubercle. The D2-receptor de-creases cAMP, blocks certain
calcium channels, and opens certain potassium channels.
Norepinephrine
Most central noradrenergic
neurons are located in the nucleus locus ceruleus of the pons and in neurons of
the reticular formation. Fibers from these nuclei inner-vate a large number of
cortical, subcortical, and spino-medullary fields. Many functions have been
ascribed to the central noradrenergic neurons, including a role in affective
disorders , in learning and memory, and in sleep–wake cycle regulation. The
mam-malian CNS contains both α- and β-adrenoceptors.
Epinephrine
Epinephrine is found only in
very low concentrations in the mammalian CNS, and it is unlikely to play a
major role as a neurotransmitter.
Serotonin
Serotonin
(5-hydroxytryptamine, or 5HT) is present in the brain as well as in the
periphery. In humans, about 90% of the total serotonin in the body is in
enterochro-maffin cells in the gastrointestinal tract; the remaining 10% occurs
primarily in the platelets and brain. The physiological significance of the
vast amounts of sero-tonin constantly synthesized and metabolized in the
pe-riphery still remains an enigma. Brain serotonin has been implicated as a
potential neurotransmitter in the media-tion of a wide variety of phenomena
(see Actions).
Synthesis and Fate
Dietary tryptophan is the
source of the formation of serotonin. Enzymes and cofactors necessary for
sero-tonin synthesis are present in both the enterochromaf-fin cells of the
gastrointestinal tract and neurons in the brain. Tryptophan is initially
hydroxylated to form 5-hydroxytryptophan. Decarboxylation of the latter
com-pound results in the formation of serotonin (Fig. 24.2).
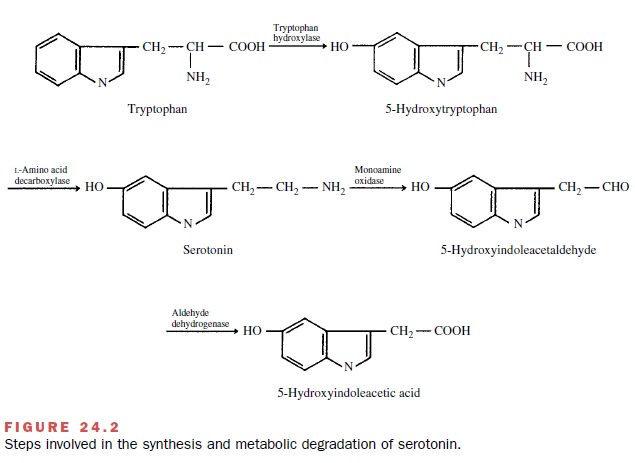
The enzymes responsible for
the metabolism of sero-tonin are present in all of the cells containing this
amine and in the liver. Serotonin is initially oxidatively deami-nated to form
5-hydroxyindoleacetaldehyde; this com-pound is subsequently rapidly oxidized to
the major metabolite 5-hydroxyindoleacetic acid, which is excreted in the
urine. Much of the serotonin released in the brain at synapses is taken back
into the initial neuron by an ac-tive reuptake mechanism to be released again.
Actions and Site of Actions
Most of the serotonin in the
brain is in the brainstem, specifically in the raphe nuclei; considerable
amounts also are present in areas of the hypothalamus, the lim-bic system, and
the pituitary gland. Current evidence in-dicates that serotonin is involved in
the regulation of several aspects of behavior, including sleep, pain
per-ception, depression, sexual activity, and aggressiveness. Some of the most
important antidepressant agents are believed to prevent the reuptake of
serotonin . Serotonin also may be involved in temper-ature regulation and in
the hypothalamic control of the release of pituitary hormones.
In addition to its presumed role as a neurotransmit-ter within the brain, serotonin is synthesized in the pineal gland, where it is a precursor for the synthesis of melatonin, a hormone that influences endocrine activ-ity, presumably by an action within the hypothalamus.
The mammalian brain appears to have an abun-dance of sites with which serotonin interacts. Fourteen distinct mammalian receptor
subtypes for serotonin have been established, not all of which have been
iden-tified in the brain. They are characterized as 5-HT1, 5-HT2,
. . . 5-HT7 subsets. There are at least five subtypes of the 5-HT1
subset and three receptor subtypes for the 5-HT2 subset.
Amino Acid Neurotransmitters
A large number of amino acids
serve as neurotransmit-ters in the mammalian CNS.
γ-Aminobutyric Acid
γ-Aminobutyric acid (GABA) is
the major inhibitory neurotransmitter in the mammalian CNS. GABA is pri-marily
synthesized (Fig. 24.3) from glutamate by the enzyme L-glutamic acid-l-decarboxylase (GAD); it is
subsequently transaminated with α-ketoglutarate by GABAA-oxoglutarate
transaminase (GABA-T) to yield glutamate and succinic semialdehyde.
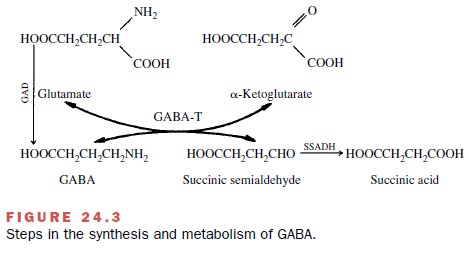
Two types of GABA receptors
have been identified in mammals, a GABAA- and a GABAB-receptor.
The GABAA-receptor (or recognition site), when coupled with GABA,
induces a shift in membrane permeability, primarily to chloride ions, causing
hyperpolarization of the neuron. This GABA receptor appears to be part of a
macromolecule that contains, in addition to the GABAA-receptor,
benzodiazepine and barbiturate binding sites and the chloride ionophore
(chloride channel). See Figure 24.4.
A number of drugs are thought
to exert their CNS effect by altering GABAA-receptor activity. The
1,4-benzodiazepines, -carbolines, barbiturates, alcohols, and general
anesthetics appear to facilitate GABA transmission by interacting at this
macromolecular complex. Vigabatrin, a newly approved anticonvulsant, elevates
brain GABA by inhibiting the breakdown en-zyme GABA-T. Several CNS convulsants,
including bicuculline, picrotoxinin, and pentylenetetrazol, are an-tagonists at
the GABA receptor. Since GABA agonists have been shown to be anticonvulsants
and GABA an-tagonists are convulsants, there is much interest in the role of
GABA in epilepsy . The GABAB-receptor, in contrast, is not modulated
by ben-zodiazepines, is not linked to chloride movement, and is not nearly as well
characterized as is the GABAA-re-ceptor. The GABAB-receptor
is coupled to K+ channels and is activated by the antispastic agent
baclofen.
Glycine
Glycine is another inhibitory
CNS neurotransmitter. Whereas GABA is located primarily in the brain, glycine
is found predominantly in the ventral horn of the spinal cord. Relatively few
drugs are known to in-teract with glycine; the best-known example is the
con-vulsant agent strychnine, which appears to be a rela-tively specific
antagonist of glycine.
Glutamic Acid and Aspartic Acid
These two excitatory amino
acids (EAAs) are widely distributed throughout the mammalian CNS. Their
ad-ministration leads to rapid depolarization of neurons and an increase in
firing rate. There are two distinct classes of EAA receptors: ionotropic
receptors and metabotropic receptors. The ionotropic receptors di-rectly gate
ion channels, while the metabotropic recep-tors are coupled to intracellular G
proteins. Receptors are named according to their sensitivity to the action of
selective agonists (Table 24.1). The best-character-ized receptor is known as
the NMDA (N-methyl-D-aspartate) receptor, which
directly gates a Mg cation channel that is also permeable to Ca++ and
NA+ . Com-pounds that block the NMDA receptor complex may attenuate
the neuronal damage following anoxia, such as occurs during a stroke; much of
the neuronal damage associated with strokes may be related to the release of
glutamic acid, aspartic acid, or both. Similarly, neuronal damage may occur as
a result of seizures, and this also may be related to excessive EAA release.
Antagonists of the NMDA receptor complex are being studied for possible uses in
strokes and other types of hypoxia.

Histamine
Histamine occurs in the
brain, particularly in certain hypothalamic neurons, and evidence is strong
that his-tamine is a neurotransmitter. Distribution of histamine, its synthetic
enzyme (histidine decarboxylase), and methyl histamine (the major brain
metabolite) is not uniform. Possible roles for histamine in the regulation of
food and water intake, thermoregulation, hormone release, and sleep have been
suggested.
Other Possible Amino Acid Neurotransmitters
Several additional amino
acids are considered to be neurotransmitter candidates. Among these are
taurine, - and -alanine, 2-phenylethylamine, and imidazole-4-acetic acid. No
available drugs are known to act via these amino acids.
Peptides as Neurotransmitters
A large number of endogenous peptides are produced by neurons that appear to possess the essential charac-teristics of neurotransmitters (e.g., their release is Ca++ dependent, they are localized in specific neurons, and their release induces changes in postsynaptic neu-ronal systems).
The names of the agents can be terribly misleading to the beginning student. Many of the peptides have been around for many years
and were named according to their known effects when they were discovered.
Examples are gastrin and cholecystokinin (CCK), com-pounds that were
historically known as gut hormones. It is important, therefore, to realize that
the names of the neuroactive peptides may bear no resemblance to their function
in the brain. Many of the neuroactive peptides exist as families of chemically
related compounds or oc-cur within larger precursor molecules (or propeptides).
However, several forms may be “active,” and several slightly different
structures may confer subtle changes in selectivity. Many neuroactive peptides
appear to co-exist and be released along with one or more of the “traditional”
neurotransmitters, such as ACh, dop-amine, or serotonin.
More than two dozen peptides
are being studied as probable central neurotransmitters, and likely many more
compounds remain to be discovered.
Substance P
The first neuropeptide to be isolated and characterized is known as substance P. Although this 11–amino acid peptide (undecapeptide) has been known for more than 60 years, its exact physiological role is still not clear. Substance P occurs in high concentrations in neurons projecting into the substantia gelatinosa layer of the spinal cord from dorsal root ganglia, among many other areas of the brain. Substance P can directly depolarize motor neurons in a manner analogous to that of other excitatory neurotransmitters. It is probable that sub-stance P is released from small unmyelinated nerve fibers in response to painful stimulation. Levels of substance P in the substantia nigra are markedly reduced in the neurological disease Huntington’s chorea.
Vasopressin and Oxytocin
Historically vasopressin and
oxytocin, two nonapep-tides, were the first peptide “neurohormones” to be
con-sidered; they are stored in the neurohypophysis and re-leased into the
bloodstream upon an appropriate stimulus. In the periphery, oxytocin stimulates
the con-traction of epididymal and uterine smooth muscle and vasopressin (antidiuretic hormone)
fa-cilitates the reabsorption of water from the kidney tubules. In addition to
these well-accepted roles as neu-rohormones, there is convincing evidence that
these compounds function as neurotransmitters; they both possess potent
inhibitory actions on neurohypophyseal neurons. The significance of their
neurotransmitter function is not yet clear.
Endogenous Opioid Peptides
A seminal discovery during
the 1960s and 1970s was the presence of endogenous substances in mammalian
brain that appeared to possess the pharmacological qualities of morphine and
other opioid analgesics. It had been known for quite awhile that most “drug
re-ceptors” were in fact receptors for endogenous trans-mitters. It was
surprising, therefore, when tissue from mouse brain was shown to avidly bind
opioids, such as morphine and heroin, in a stereoselective manner. As Avram
Goldstein, one of the pharmacologists in-volved in discovering the endogenous
opioids, noted, “It seemed unlikely, a
priori, that such highly stereo-specific receptors should have been
developed by na-ture to interact with alkaloids from the opium poppy.”1
A series of peptides,
occurring naturally in brain and possessing pharmacological properties similar
to those of morphine, have been described. At least three separate families of
peptides have opioid prop-erties (Table 24.2), and the different classes of
pep-tides reside in separate distinct neurons. It is likely that the endogenous
opioid peptides coexist in neu-rons with other nonopioid neurotransmitters. The
ini-tial hope that these endogenous agents or synthetic derivatives of them
would be found to retain the anal-gesic activity of the opioids but be devoid
of respira-tory depression and/or addictive properties has now somewhat abated.

Related Topics