Chapter: Essential Clinical Immunology: Immune-Mediated Neurological Syndromes
Central Nervous System Systemic Lupus Erythematosus
CENTRAL NERVOUS SYSTEM SYSTEMIC
LUPUS ERYTHEMATOSUS
CNS SLE is an example of an autoim-mune pathology
with abnormalities also detected in other organs outside the cen-tral or
peripheral nervous system. Over ten years’ follow-up, approximately 25 percent
of SLE patients will develop CNS manifes-tations.
One hypothesis is that the neuropsy-chiatric
symptoms of SLE may result from secondary causes. Infarctions may be
experienced by patients, felt to be related to antibodies found in SLE, which
affect the clotting system, such as circulating lupus anticoagulant or antiphospholipid
antibodies (APLAs). Another mechanism may be symptoms secondary to CNS
infec-tions such as meningitis or encephalitis. Treatments of SLE require
immunosup-pressive medications or steroids, which may predispose patients to
these etiolo-gies. Metabolic issues may also present as neuropsychiatric
symptoms, such as in the case of uremia secondary to renal involve-ment of SLE.
Lupus
cerebritis is the term used to con-vey that
the CNS symptomatology is due to the SLE immune process. This broad etiology of
neuropsychiatric symptoms may result from a demyelinating pathol-ogy. The
symptoms associated with CNS SLE may be diffuse in nature, rather than a focal
symptom suggestive of a vascu-lar territory and therefore stroke. These
symptoms may include aseptic meningi-tis, headache, chorea, myelopathy, cranial
neuropathy, seizures, confusion, and cog-nitive dysfunction. Also felt to be
due to
MRI scanning is the clinical tool of choice in
investigating the diagnosis of CNS SLE. Infarctions can be identified, as well
as diffuse demyelinating disease. Some lesions may resolve on subsequent MRI
testing and therefore point away from infarction as the etiology of the
clinical manifestation. Infarctions are permanent, whereas demyelination may be
reversible. In fact, clinical improvement has been cor-related with resolving
lesions, following immunosuppressive therapy on MRI scan-ning, and resolving
lesions may actually predict an improved clinical course.
Diffuse CNS SLE is difficult to diagnose and
manage. CSF studies, anti-DNA anti-body titers, complement levels, and even
imaging may be nonspecific or insensitive. For instance, if a psychosis
develops in the SLE patient, it may be due to the SLE, or the use of steroids,
a mainstay of the phar-macopoeia in this disease. Depression may also be a
manifestation of lupus cerebritis or may be a reactive depression to having the
disease. However, missing the diagno-sis of lupus cerebritis or mistreating it
car-ries high mortality and morbidity.
In 1986, the first reports of an antibody
population to ribosomal P proteins and association to lupus cerebritis came to
the literature. This work was based on earlier findings that patients produced
antibod-ies that bound to ribosomes. The antigens were found to be
phosphoproteins or “P proteins” located on the 60S subunit of ribosomes. These
three proteins were labeled P0 (35 kD molecular weight), P1 (19 kD), and P2 (17
kD). These proteins are felt to be involved in protein synthesis; monoclonal
antibodies to these proteins inhibit the elongation factors EF-1 and EF-2 to
ribosomes and inhibit protein synthe-sis. In addition to being on ribosomes,
the P proteins are also on the cell surfaces of diffuse etiology are
psychiatric symptoms, such as mood disorders and psychosis. Other neurological
symptoms outside the CNS include peripheral neuropathy or mononeuritis
multiplex.
The combination of pathological stud-ies and
positron emission tomography scanning form a hypothesis: hypoperfu-sion causes
a breakdown of the blood-brain barrier. The loss of integrity allows
antineuronal antibodies to cross over and ultimately lead to demyelination,
similar to theories in MS. The inciting factors that would cause hypoperfusion
are unknown. Evidence of a vasculopathy is present in pathological specimens,
which dem-onstrate a perivascular accumulation of mononuclear cells, without
destruction of the blood vessels. There may also be small infarctions secondary
to occlusion of the vessel lumen. APLAs are associated with stroke syndromes
and may play a role in the vasculopathy of CNS SLE.
The effects of these APLAs may include a
prothrombotic state with coagu-lation pathway abnormalities and cause
thromboembolism, stroke, focal seizures, migraines, and multi-infarct dementia.
In this setting, the treatment of the disease would be directed more at the
antithrom-botic state, with aspirin and consideration of anticoagulation,
rather than with ste-roids or immunosuppressants.
Steroids are used only if there is coexist-ing
serological evidence of active SLE. Ste-roids may be detrimental in a pure
stroke setting, without evidence of active immune disease. Clinically, CNS
diffuse vasculitis presents as fever, severe headaches, and confusion, with
symptoms that progress rapidly to psychosis, seizures, or coma. Serologically
active SLE is detected with elevated anti-dsDNA (double-stranded DNA) and
hypocomplementemia.
neuronal, hepatoma, and activated T cells. ELISA
testing is the assay of choice against purified human ribosomal P proteins.
This antibody is specific to SLE when tested
against other autoimmune dis-eases, such as rheumatoid arthritis, sclero-derma,
and myositis, but the incidence rate is variable among different studies of SLE
patients. The antibody is especially linked to patients with lupus-related
psy-chosis (18/20 patients) and severe depres-sion (88 percent). Others in the
literature have not been able to confirm these results. Small patient numbers
confound the con-clusions of many of these studies.
Ribosomal P protein–targeted antibod-ies do not
appear to be synthesized in the CNS and are much more prevalent in the serum of
SLE patients. One possible expla-nation is that the antibodies are cell bound
in the CNS, as normal neuronal tissue may also express the ribosomal P
proteins. Overall, these antibodies might be consid-ered specific for CNS SLE,
although not very sensitive. They are present indepen-dently of other
SLE-associated antibodies such as to dsDNA and may be helpful in diagnosis if
the other marker antibodies have returned to normal.
Other antibodies are being investigated for
specificity for CNS SLE. An antineuro-nal antibody was produced against human
neuroblastoma cell lines. These were detected in 45 percent of patients with
CNS SLE, but on 5 percent of those with SLE but without CNS involvement.
Lympho-cytotoxic antibodies have been associated with decreased cognitive
function. Cross-reactive antibodies have been found in the sera and CSF of SLE
patients that bind to double stranded DNA and excitatory N-methyl-D-aspartate
(NMDA) receptors on neurons. Still it is not known if all of these antibody
populations are causative
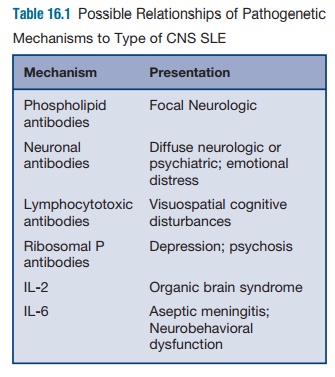
Neurological manifestations can be divided into
diffuse symptoms and focal symptoms, which may be correlated with different
immune event (Table 16.1). It is generally believed that diffuse symptoms, such
as seizures, psychosis, or mental status changes may be associated with
anti-lymphocyte antibodies cross-reac-tive with neuronal antigens. Autoimmune
antibodies can be directed against neu-rofilaments, phospholipids, glycolipids,
glycoproteins, and neuronal cell surface antigens. Widely studied autoantigens
are the 32-kD cell surface antigen, the 50–52-kD cell surface antigen, and the
97–98 kD antigen. Patients with lympho-cytotoxic antibodies have been shown to
have visuospatial abnormalities and verbal deficits on neuropsychological
testing, not detected in those without these antigens. In this setting, the
treatment would aim for anti-inflammatory and immunosup-pressive mechanisms,
using steroids and possibly cyclophosphamide and plasma-pheresis. Meningitis
may present as well and may be secondary to bacterial infec-tions due to
immunosuppression. In addi-tion, aseptic meningitis may be caused by
medications, such as ibuprofen and immu-nosuppressive medication.
Partial or generalized seizures may also occur in
the setting of SLE and gen-erally portend a poor prognosis. Patients are
generally treated with anticonvul-sants, and steroids are only added if it is
felt that the patient is in an active flare, to prevent potential permanent
scarring and a seizure focus.
Although migraine headaches occur frequently in the
setting of SLE, a causative effect has not been determined. More seri-ously,
patients may have other types of headache, such as pseudotumor cerebri,
cerebral venous, or sagittal sinus throm-bosis. This is usually due to a
secondary cause of SLE, such as renal dysfunction and a hypercoagulable state.
The sudden development of headaches in a previously headache-free patient or
neurological symptoms or signs requires rapid work-up and imaging.
More uncommon CNS syndromes occur and may be
varied. Movement disorders such as chorea or ataxia point to disease in the
basal ganglia or cerebellum. These dis-orders are usually self-limited to two
to six weeks and may not require therapy. If they are associated with APLA,
anticoagulation may be considered.
Patients may have cranial neuropa-thies causing
symptoms such as double vision or hearing loss, trigeminal neural-gia, or
dysarthria. Steroids are generally used in these conditions, and if refractory,
cyclophosphamide, an immunosuppres-sant medication, is used. When patients
present with these symptoms, the question of MS should be ruled out. Both
diseases may have optic neuritis, transverse myelitis, or inflammation of the
spinal cord (which must be treated aggressively with, at the very least,
steroids, and possibly plasma-pheresis and cyclophosphamide for recov-ery), or
internuclear ophthalmoplegia, and may have similar MRI scans. Patients who do
have similar scans and who have lupus are referred to as having “lupus
sclerosis.” APLA in this setting often help with dif-ferentiation of the
syndromes. MS should have no peripheral manifestations out-side of the CNS.
Cytokine release may play a role in these
pathologies. IL-1, IL-2, IL-6, TNF-α, IFN-α, and IFN-γ have all
been demon-strated to be released by CNS microglial cells and astrocytes by LPS
stimulation. In the CNS, a cascade of pro-inflammatory cytokines causes events
of inflammation similar to peripheral macrophages and monocytes. These
cytokines not only medi-ate inflammation but also may directly affect brain
function, as there are cytokine receptors on the hypothalamus. Antibod-ies can
cause functional dysregulation by blocking the release of neurotransmitters and
neuropeptides, resulting in abnormal electrophysiological testing, such as the
electroencephalogram.
Animal models have been used to investigate the
human disease with mixed success. The MRL/P mice have neurobe-havioral
abnormalities such as timidity, phobic behavior, and anxiety. This mani-fests
at age 7 to 8 weeks, when autoanti-bodies to brain tissue also present. The
pathology demonstrates B lymphocytes around the choroid plexus with MHC class
II antigen presentation. This differs from the vasculitis that is more typical
of the human SLE syndrome. However, like humans with an exacerbation of SLE,
IL-6 is elevated in the CSF and the animal behavior can be significantly
improved by treating with immunosuppression and steroids. The clinical
improvement is cor-related with a decrease in IL-6 levels in the CSF.
The SLE syndrome of focal events sec-ondary to APLA
can also be demonstrated in a mouse model by injecting MRL/pr mice with APLA
and anti-neuronal anti-bodies. Antigen derived from limbic brain structures,
which also react to human sera, the B2 glycoprotein, causes production of APLA
and ischemic events/stokes.
The future of CNS SLE may lie in stem cell
transplant. One study of seven patients using chemotherapy followed by
autolo-gous stem cell transplantation yielded excellent clinical results at a
median follow-up of twenty-five months. The patients had initially suffered
from cerebritis, myelitis, vasculitis, or glomerulonephritis and were
unresponsive to multiple courses of cyclo-phosphamide. At follow-up, all patients
were free of signs of active lupus and sero-logical markers had improved. The
theory of the treatment is that when memory T cells are removed from influence,
maturation of new lymphocyte progenitor cells occurs without recruiting
autoimmune activity.
Related Topics