Chapter: Pathology: Genetic Disorders
Autosomal Recessive Disorders
AUTOSOMAL RECESSIVE DISORDERS
(This
material is included here for reinforcement. It is also covered in the
Physiol-ogy Lecture Notes.)
Cystic
fibrosis (CF) is the most common lethal genetic disorder in Caucasians.
Itis due to mutation of the chloride channel protein, cystic fibrosis
transmembrane conductance regulator (CFTR), whose CFTR gene is located on chromosome 7 and most commonly has been
damaged by a deletion of the amino acid phenylalanine at position 508 ( F508).
The defective chloride channel protein leads to abnormally thick viscous mucus,
which obstructs the ducts of exocrine organs.
The
distribution of disease reflects the
distribution of eccrine sweat glands and exocrine glands.
·
In the lungs, CF may cause recurrent
pulmonary infections; chronic bronchi-tis; and bronchiectasis.
·
In the pancreas, CF may cause
plugging of pancreatic ducts resulting in atro-phy and fibrosis; and pancreatic
insufficiency leading to fat malabsorption, malodorous steatorrhea, and
deficiency of fat-soluble vitamins.
·
In the male reproductive system, CF
may be associated with absence or obstruction of the vas deferens and
epididymis, which often leads to male infertility.
·
In the liver, plugging of the
biliary canaliculi may result in biliary cirrhosis.
·
In the GI tract, the thick
secretions may cause small intestinal obstruction (meconium ileus).
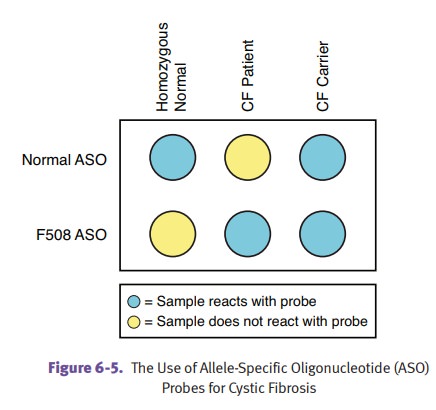
Diagnosis
can be established with a sweat test (elevated NaCl) or DNA probes. Due to
improved therapies, some patients live into their forties, but with this increase
in longevity there has been an increase in liver disease. Patients succumb to
pulmonary disease. The 3 most common pulmonary infections are S. aureus, H. influenzae, and P. aeruginosa . Lung transplantation is
a treatment option. Patients infected with Burkholderia
cepacia complex who undergo transplant have a worse prognosis.
Phenylketonuria
(PKU) is due to deficiency of phenylalanine hydroxylase,
resultingin toxic levels of phenylalanine and a lack of tyrosine.
Clinically,
affected children are normal at birth but, if undiagnosed and untreated,
develop intellectual development disorder by age 6 months. The lack of tyrosine
causes light-colored skin and hair, since melanin is a tyrosine derivative.
Affected children may have a mousy or musty odor to the sweat and urine
(secondary to metabolite [phenylacetate] accumulation).
Screening
for PKU is done at birth. Treatment is dietary restriction of phenylalanine,
including avoidance of the artificial sweetener aspartame.
A
genetic variant, benign
hyperphenylalaninemia, has partial enzyme deficiency with mildly increased
levels of phenylalanine which are insufficient to cause intel-lectual
disability.
In
a minority of cases, an abnormality of the cofactor tetrahydrobiopterin causes
a variant that does not respond to dietary restriction.
Transplacental
accumulation of phenylalanine can cause problems with fetal devel-opment in
cases of maternal PKU. Prevention requires maternal dietary restriction.
Alkaptonuria
(ochronosis) occurs when deficiency of homogentisic acid
oxidaseresults in the accumulation of homogentisic acid. The homogentisic acid
has an affinity for connective tissues (especially cartilage), resulting in a
black discoloration (as a consequence of oxidation of homogentisic acid).
Clinical
features include urine that is initially pale yellow but turns black upon
standing, and black-stained cartilage, which causes discoloration of the nose
and ears. Alkaptonuria also predisposes for early onset of degenerative
arthritis.
Albinism is
caused by a lack of the enzyme tyrosinase needed for melanin produc-tion.
Affected individuals show deficiency of melanin pigmentation in the skin, hair
follicles, and eyes (oculocutaneous albinism), with resulting increased risk of
basal cell and squamous cell carcinomas.
The
glycogen
storage diseases are a group of rare diseases that have in common a
deficiency of one of the enzymes necessary for the metabolism of glycogen,
which results in the accumulation of glycogen in the liver, heart, and skeletal
muscle.
·
Type I (von Gierke disease) is due to
a deficiency ofglucose-6-phosphatase,
andis characterized clinically by hepatomegaly and hypoglycemia.
·
Type II (Pompe disease) is due to
a deficiency oflysosomalα-1,4-glucosidase(acid
maltase), and is characterized clinically by hepatomegaly, skeletal
musclehypotonia, cardiomegaly, and death from cardiac failure by age 2 years.
·
Type V (McArdle syndrome) is due to
a deficiency ofmuscle glycogen
phos-phorylase, and is characterized clinically by exercise-induced muscle
cramps.
Tay-Sachs
disease is due to a deficiency of hexosaminidase A (due to mutation
ofHEXA gene on chromosome 15), which
leads to the accumulation of GM2 ganglio-side in the lysosomes of the CNS and
retina. Tay-Sachs is common in Ashkenazi Jews (1 in 30 carrier rate).
The
distribution of disease involves the retina (cherry-red spot due to
accentua-tion of the macula) and central nervous system (dilated neurons with
cytoplasmic vacuoles). Affected children are normal at birth, but by 6 months
show onset of symptoms (progressive mental deterioration and motor
incoordination) that prog-ress to death by age 2–3 years. Electron microscopy shows distended lysosomes with whorled
membranes; the diagnosis can also be established with enzyme assays and DNA
probes.
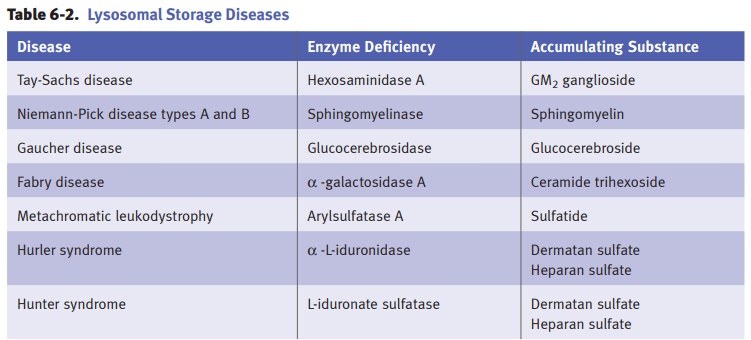
Niemann-Pick
disease is caused by a deficiency of sphingomyelinase, which leadsto
the accumulation of sphingomyelin within the lysosomes of the CNS and reticu-loendothelial
system (monocytes and macrophages located in reticular connective tissue).
Niemann-Pick is common in Ashkenazi Jews (note similarity to Tay-Sachs
disease).
The
distribution of disease depends on the form of disease, but can involve the
retina (cherry-red spot, note similarity to Tay-Sachs disease); central nervous
system (dis-tended neurons with a foamy cytoplasmic vacuolization, note
similarity to Tay-Sachs disease); and reticuloendothelial system
(hepatosplenomegaly, lymphadenopathy, and bone marrow involvement; note
difference from Tay-Sachs disease).
In
Neimann-Pick types A and B , there
is a mutation affecting an enzyme that metabolizes lipids; organomegaly occurs,
and with type A, there is severe neuro-logic damage. In type C—the most common form—a defect in cholesterol transport
causes ataxia, dysarthria, and learning difficulties. All forms are lethal,
usually before adulthood.
Gaucher
disease is the most common lysosomal storage disorder. Deficiency
ofglucocerebrosidase leads to the accumulation of glucocerebroside,
predominately in the lysosomes of the reticuloendothelial system (monocytes and
macrophages located in reticular connective tissue).
Type I represents
99% of cases and presents in adulthood with hepatosplenomeg-aly; thrombocytopenia/pancytopenia
secondary to hypersplenism; lymphadenopa-thy; and bone marrow involvement that
may lead to bone pain, deformities, and fractures. Central nervous system
manifestations occur in types II and III.
The
characteristic Gaucher cells are enlarged
macrophages with a fibrillary (tissue paper–like) cytoplasm. Diagnosis can be
established with biochemical enzyme assay of glucocerebrosidase activity.
Mucopolysaccharidosis
(MPS) is a group of lysosomal storage disorders
charac-terized by deficiencies in the lysosomal enzymes required for the
degradation of mucopolysaccharides (glycosaminoglycans).
Clinical
features include intellectual disability; cloudy cornea; hepatosplenomeg-aly;
skeletal deformities and coarse facial features; joint abnormalities; and
cardiac lesions. MPS I (Hurler syndrome) is the severe form and is due to
deficiency of α-L-iduronidase.
MPS II (Hunter syndrome) is a milder form; it shows X-linkedrecessive
inheritance and is due to a deficiency of L-iduronate sulfatase.
Related Topics